- Introduction to Genomics
- Educational Resources
- Policy Issues in Genomics
- The Human Genome Project
- Funding Opportunities
- Funded Programs & Projects
- Division and Program Directors
- Scientific Program Analysts
- Contact by Research Area
- News & Events
- Research Areas
- Research investigators
- Research Projects
- Clinical Research
- Data Tools & Resources
- Genomics & Medicine
- Family Health History
- For Patients & Families
- For Health Professionals
- Jobs at NHGRI
- Training at NHGRI
- Funding for Research Training
- Professional Development Programs
- NHGRI Culture
- Social Media
- Broadcast Media
- Image Gallery
- Press Resources
- Organization
- NHGRI Director
- Mission & Vision
- Policies & Guidance
- Institute Advisors
- Strategic Vision
- Leadership Initiatives
- Diversity, Equity, and Inclusion
- Partner with NHGRI
- Staff Search

About Alpha-1 Antitrypsin Deficiency
Alpha-1 antitrypsin deficiency is an inherited condition that causes low levels of, or no, alpha-1 antitrypsin in the blood.
What is alpha-1 antitrypsin deficiency?
Alpha-1 antitrypsin deficiency (AATD) is an inherited condition that causes low levels of, or no, alpha-1 antitrypsin (AAT) in the blood. AATD occurs in approximately 1 in 2,500 individuals. This condition is found in all ethnic groups; however, it occurs most often in whites of European ancestry.
Alpha-1 antitrypsin (AAT) is a protein that is made in the liver. The liver releases this protein into the bloodstream. AAT protects the lungs so they can work normally. Without enough AAT, the lungs can be damaged, and this damage may make breathing difficult.
Everyone has two copies of the gene for AAT and receives one copy of the gene from each parent. Most people have two normal copies of the alpha-1 antitrypsin gene. Individuals with AATD have one normal copy and one damaged copy, or they have two damaged copies. Most individuals who have one normal gene can produce enough alpha-1 antitripsin to live healthy lives, especially if they do not smoke.
People who have two damaged copies of the gene are not able to produce enough alpha- 1 antitrypsin, which leads them to have more severe symptoms.
What are the symptoms of AATD?
Alpha-1 antitrypsin deficiency (AATD) can present as lung disease in adults and can be associated with liver disease in a small portion of affected children. In affected adults, the first symptoms of AATD are shortness of breath with mild activity, reduced ability to exercise and wheezing. These symptoms usually appear between the ages of 20 and 40. Other signs and symptoms can include repeated respiratory infections, fatigue, rapid heartbeat upon standing, vision problems and unintentional weight loss.
Some Individuals with AATD have advanced lung disease and have emphysema, in which the small air sacs (alveoli) in the lungs are damaged. Symptoms of emphysema include difficulty breathing, a hacking cough and a barrel-shaped chest. Smoking or exposure to tobacco smoke increases the appearance of symptoms and damage to the lungs. Other common diagnoses include COPD (chronic obstructive pulmonary disease), asthma, chronic bronchitis and bronchiectasis - a chronic inflammatory or degenerative condition of one or more bronchi or bronchioles.
Liver disease, called cirrhosis of the liver, is another symptom of AATD. It can be present in some affected children, about 10 percent, and has also been reported in 15 percent of adults with AATD. In its late stages signs and symptoms of liver disease can include a swollen abdomen, coughing up blood, swollen feet or legs, and yellowing of the skin and the whites of the eyes (jaundice).
Rarely, AATD can cause a skin condition known as panniculitis, which is characterized by hardened skin with painful lumps or patches. Panniculitis varies in severity and can occur at any age.
How is AATD diagnosed?
Alpha-1 antitrypsin deficiency (AATD) is diagnosed through testing of a blood sample, when a person is suspected of having AATD. For example, AATD may be suspected when a physical examination reveals a barrel-shaped chest, or, when listening to the chest with a stethoscope, wheezing, crackles or decreased breath sounds are heard.
Testing for AATD, using a blood sample from the individual, is simple, quick and highly accurate.. Three types of tests are usually done on the blood sample:
Alpha-1 genotyping, which examines a person's genes and determines their genotype.
Alpha-1 antitrypsin PI type of phenotype test, which determines the type of AAT protein that a person has.
Alpha-1 antitrypsin level test, which determines the amount of AAT in a person's blood.
Individuals who have symptoms that suggest AATD or who have a family history of AATD should consider being tested.
What is the treatment for AATD?
Treatment of alpha-1 antitrypsin deficiency (AATD) is based on a person's symptoms. There is currently no cure. The major goal of AATD management is preventing or slowing the progression of lung disease.
Treatments include bronchodilators and prompt treatment with antibiotics for upper respiratory tract infections. Lung transplantation may be an option for those who develop end-stage lung disease. Quitting smoking, if a person with AATD smokes, is essential.
Replacement (augmentation) therapy with the missing AAT protein is available, although it is used only under special circumstances. It is not known how effective this is once disease has developed or which people would benefit most.
Is AATD inherited?
Alpha-1 antitrypsin deficiency (AATD) is inherited in families in an autosomal codominant pattern. Codominant inheritance means that two different variants of the gene (alleles) may be expressed, and both versions contribute to the genetic trait.
The M gene is the most common allele of the alpha-1 gene. It produces normal levels of the alpha-1 antitrypsin protein.
The Z gene is the most common variant of the gene. It causes alpha-1 antitrypsin deficiency. The S allele is another, less common variant that causes ATTD.
If a person inherits one M gene and one Z gene or one S gene ('type PiMZ' or 'type PiMS'), that person is a carrier of the disorder. While such a person may not have normal levels of alpha-1 antitrypsin, there should be enough to protect the lungs. However, carriers with the MZ alleles have an increased risk for lung disease, particularly if they smoke.
A person who inherits the Z gene from each parent is called 'type PiZZ.' This person has very low alpha-1 antitrypsin levels, allowing elastase - an enzyme especially of pancreatic juice that digests elastin - to damage the lungs. A person who inherits an altered version called S and Z is also likely to develop AATD.
Additional Resources for Alpha-1 anttrypsin deficiency.
Alpha1 -Antitrypsin Deficiency Registry
Alpha-1 antitrypsin deficiency - Genetics Home Reference
Alpha-1 Antitrypsin Deficiency - National Library of Medicine
Alpha-1 Association Genetic Counseling Service
Alpha-1 Advocacy Alliance
Alpha 1-Antitrypsin Deficiency
Last updated: January 4, 2012

An official website of the United States government
Here’s how you know
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( A locked padlock ) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.
- Heart-Healthy Living
- High Blood Pressure
- Sickle Cell Disease
- Sleep Apnea
- Information & Resources on COVID-19
- The Heart Truth®
- Learn More Breathe Better®
- Blood Diseases and Disorders Education Program
- Publications and Resources
- Blood Disorders and Blood Safety
- Sleep Science and Sleep Disorders
- Lung Diseases
- Health Disparities and Inequities
- Heart and Vascular Diseases
- Precision Medicine Activities
- Obesity, Nutrition, and Physical Activity
- Population and Epidemiology Studies
- Women’s Health
- Research Topics
- Clinical Trials
- All Science A-Z
- Grants and Training Home
- Policies and Guidelines
- Funding Opportunities and Contacts
- Training and Career Development
- Email Alerts
- NHLBI in the Press
- Research Features
- Past Events
- Upcoming Events
- Mission and Strategic Vision
- Divisions, Offices and Centers
- Advisory Committees
- Budget and Legislative Information
- Jobs and Working at the NHLBI
- Contact and FAQs
- NIH Sleep Research Plan
- Health Topics
- < Back To COPD
- Alpha-1 Antitrypsin Deficiency
- What Is COPD?
- Causes and Risk Factors
- Living With
MORE INFORMATION
COPD Alpha-1 Antitrypsin Deficiency
Language switcher, what is alpha-1 antitrypsin deficiency.
Alpha-1 antitrypsin (AAT) deficiency is a condition that raises your risk for lung and other diseases.
AAT is a protein made in your liver to help protect the lungs. If your body does not make enough AAT, your lungs are more easily damaged from smoking, pollution, or dust from the environment. This can lead to COPD or bronchiectasis , another lung disease. AAT deficiency may also cause liver disease. The liver disease can occur among infants and children, and the lung disease usually occurs in individuals who are older than 30.
AAT deficiency runs in families. Many people do not know that they have it, but early diagnosis can help prevent COPD and other serious lung diseases. Talk to your healthcare provider if you have a family member who has AAT deficiency or who was a smoker diagnosed with COPD between ages 40 and 50. Also, talk to your provider if you have symptoms such as an ongoing cough, shortness of breath, wheezing, or liver disease.
Another important step to prevent or delay COPD is to quit smoking. If you do not smoke, do not start.
How do you get it?
Because AAT deficiency is an inherited disease, meaning it runs in families, it cannot be prevented. It can happen to anyone of any race or ethnicity. However, it is more common in white people of Northern European backgrounds.
Everyone inherits two AAT genes, one gene from each parent. If you inherit a mutated or changed gene from each parent, you will have AAT deficiency.
If you inherit a mutated AAT gene from one parent and a normal AAT gene from the other parent, you are a carrier for the condition. You might have lower levels of AAT protein in your blood, but you most likely will not have AAT deficiency. You might also pass the mutated gene to your children.

AAT deficiency is a complex disease, and many factors — some known, like smoking, and others still unknown — contribute to how it affects different people. Sometimes, even if you inherit two mutated AAT genes, you may not have any symptoms or health problems. You may never even realize that you have AAT deficiency.
You may want to talk to a genetic counselor if you are planning to have children and think they are at risk of having AAT deficiency. A genetic counselor can answer questions about the risk and explain the choices that are available.
What are the symptoms?
Some people do not have any symptoms. For those who do, symptoms usually appear in people between 20 and 50 years old.
Often, people are diagnosed with asthma first. This is because wheezing is also a symptom of asthma. Also, people who have AAT deficiency respond well to asthma medicines. Some people can develop symptoms of COPD .
Some people who have AAT deficiency may have liver damage. Signs of liver damage include jaundice and swelling in the legs. Rarely, AAT deficiency can cause skin problems, such as painful lumps or patches.
How is it diagnosed?
Your healthcare provider may test you for AAT deficiency if you have relatives who have AAT deficiency or a lung or liver disease or after you develop a lung or liver disease that is related to the condition.
- A blood test can check the level of AAT protein in your blood. If the level is lower than normal, it is likely that you have AAT deficiency.
- A genetic test is the most certain way to check for AAT deficiency and should be done to confirm the results of the blood test and find the mutation in the AAT gene. A genetic counselor can help you understand what to expect from a genetic test and what your results mean.
- A pulmonary test to see how well your lungs are working may be recommended by your healthcare provider if you have COPD related to AAT deficiency.
How is it treated?
There is no cure for AAT deficiency, but there are treatments to slow the lung damage it causes. If you have emphysema and AAT, standard COPD treatments — bronchodilators, inhaled steroids, antibiotics, oral corticosteroids, regular vaccinations, pulmonary rehabilitation, oxygen therapy, and (for severe cases) surgery — may be given.
If you have emphysema, you may need a lifelong treatment called augmentation therapy. This treatment involves raising the AAT protein to acceptable amounts. This helps slow down lung damage. Side effects of this treatment are rare and may include a mild fever, headaches, nausea, and dizziness.
If you have COPD, you may also need medicines or other treatments . Talk to your healthcare provider about ways to help prevent or delay lung damage, such as quitting smoking and avoiding secondhand smoke, dust, or air pollution.

An official website of the United States government
The .gov means it's official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you're on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
- Browse Titles
NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

GeneReviews ® [Internet].
- GeneReviews by Title
- GeneReviews Advanced Search
Alpha-1 Antitrypsin Deficiency
James K Stoller , MD, MS, Vera Hupertz , MD, and Loutfi S Aboussouan , MD.
Initial Posting: October 27, 2006 ; Last Revision: June 1, 2023 .
Estimated reading time: 33 minutes
Clinical characteristics.
Alpha-1 antitrypsin deficiency (AATD) can present with hepatic dysfunction in individuals from infancy to adulthood and with chronic obstructive lung disease (emphysema and/or bronchiectasis), characteristically in individuals older than age 30 years. Individuals with AATD are also at increased risk for panniculitis (migratory, inflammatory, tender skin nodules which may ulcerate on legs and lower abdomen) and C-ANCA-positive vasculitis (granulomatosis with polyangiitis). Phenotypic expression varies within and between families. In adults, smoking is the major factor in accelerating the development of COPD; nonsmokers may have a normal life span, but can also develop lung and/or liver disease. Although reported, emphysema in children with AATD is extremely rare. AATD-associated liver disease, which is present in only a small portion of affected children, manifests as neonatal cholestasis. The incidence of liver disease increases with age. Liver disease in adults (manifesting as cirrhosis and fibrosis) may occur in the absence of a history of neonatal or childhood liver disease. The risk for hepatocellular carcinoma (HCC) is increased in individuals with AATD.
Diagnosis/testing.
The diagnosis of AATD relies on demonstration of low serum concentration of alpha-1 antitrypsin (AAT) and either identification of biallelic pathogenic variants in SERPINA1 or detection of a functionally deficient AAT protein variant by protease inhibitor (PI) typing. Note: The unconventional nomenclature of SERPINA1 alleles is based on electrophoretic protein variants that were identified long before the gene ( SERPINA1) was known. Alleles were named with the prefix PI* (protease inhibitor*) serving as an alias for the gene. Using this nomenclature, the most common (normal) allele is PI*M and the most common pathogenic allele is PI*Z.
Management.
Treatment of manifestations: COPD is treated with standard therapy. Augmentation therapy with periodic intravenous infusion of pooled human serum alpha-1 antitrypsin (AAT) is used in individuals who have established emphysema. Lung transplantation may be an appropriate option for individuals with end-stage lung disease. Liver transplantation is the definitive treatment for severe disease (will restore AAT levels). Dapsone or doxycycline therapy is used for panniculitis; if refractory to this, high-dose intravenous AAT augmentation therapy is indicated.
Surveillance: Every six to 12 months: pulmonary function tests including spirometry with bronchodilators and diffusing capacity measurements; liver function tests, platelet count and liver ultrasound, elastography (e.g., FibroScan), magnetic resonance imaging.
Agents/circumstances to avoid: Smoking (both active and passive); occupational exposure to environmental pollutants used in agriculture, mineral dust, gas, and fumes; excessive use of alcohol.
Evaluation of relatives at risk: Evaluation of parents, older and younger sibs, and offspring of an individual with severe AATD in order to identify as early as possible those relatives who would benefit from institution of treatment and preventive measures.
Genetic counseling.
AATD is inherited in an autosomal codominant manner. If both parents are heterozygous for one SERPINA1 pathogenic variant (e.g., PI*MZ), each sib of an affected individual has a 25% chance of being affected (PI*ZZ), a 50% chance of being heterozygous (PI*MZ), and a 25% chance of inheriting neither of the pathogenic variants (PI*MM). In the less frequent instance in which one parent is homozygous (PI*ZZ) and one parent is heterozygous (PI*MZ), the risk to each sib of being homozygous (PI*ZZ) is 50%. Unless an individual with AATD has children with an affected individual or a heterozygote , offspring will be obligate heterozygotes for a pathogenic variant. (Risk of lung disease may be increased in heterozygous individuals depending on their environmental exposures such as smoking.) Heterozygote testing for at-risk family members and prenatal and preimplantation genetic testing are possible once the pathogenic SERPINA1 variants have been identified in the family.
Suggestive Findings
Alpha-1 antitrypsin deficiency (AATD) should be suspected in individuals with evidence of:
- Chronic obstructive pulmonary disease (i.e., emphysema, persistent airflow obstruction, chronic bronchitis, and/or bronchiectasis); AND/OR
- Liver disease at any age, including obstructive jaundice in infancy
- C-ANCA positive vasculitis (i.e., granulomatosis with polyangiitis)
- Necrotizing panniculitis
Establishing the Diagnosis
The diagnosis of AATD relies on demonstration of low serum concentration of AAT AND EITHER OF THE FOLLOWING:
- Identification of SERPINA1 pathogenic variants
- Detection of a functionally deficient AAT protein
Demonstration of Low Serum Concentration of the Protein Alpha-1 Antitrypsin (AAT)
A variety of techniques have been used to measure serum AAT concentration; currently the most commonly used technique is nephelometry.
- Normal serum levels are 20-53 µmol/L or approximately 100-220 mg/dL by nephelometry.
- Serum levels observed in AATD with lung disease are usually <57 mg/dL.
Identification of Biallelic Pathogenic Variants in SERPINA1
Molecular genetic testing approaches can include a combination of gene -targeted testing (single-gene testing, multigene panel ) and comprehensive genomic testing ( exome sequencing , exome array , genome sequencing ) depending on the phenotype .
Gene-targeted testing requires that the clinician determine which gene (s) are likely involved, whereas genomic testing does not. Because the phenotype of AATD is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1 ), whereas those in whom the diagnosis of AATD has not been considered are more likely to be diagnosed using genomic testing (see Option 2 ).
Option 1. When the phenotypic and laboratory findings suggest the diagnosis of AATD molecular genetic testing approaches can include single- gene testing or use of a multigene panel :
- Single- gene testing. Targeted analysis for the common PI*Z, PI*S, PI*I, and PI*F alleles (see Molecular Genetics for standard nomenclature) may be performed first. Sequence analysis of SERPINA1 detects other pathogenic variants such as small intragenic deletions/insertions and missense , nonsense , and splice site variants. Note: Depending on the sequencing method used, single- exon , multiexon, or whole- gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes SERPINA1 and other genes of interest (see Differential Diagnosis ) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype . Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview . (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis , deletion/duplication analysis , and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1 ). For an introduction to multigene panels click here . More detailed information for clinicians ordering genetic tests can be found here .
Option 2. When the diagnosis of AATD is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene [s] are likely involved) is the best option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi) exon deletions or duplications that cannot be detected by sequence analysis .
For an introduction to comprehensive genomic testing click here . More detailed information for clinicians ordering genomic testing can be found here .
Note: The nomenclature of SERPINA1 alleles is unconventional because it is based on electrophoretic protein variants that were identified long before the gene ( SERPINA1 ) was identified [ Cox et al 1980 ]. Because this older nomenclature is well established in the literature, it is used in this GeneReview .
SERPINA1 alleles encoding the variant AAT proteins were named with the prefix PI* (protease inhibitor*) serving as an alias for SERPINA1 (which had yet to be identified). The six SERPINA1 alleles discussed here are the following. (See Molecular Genetics for more details and information on other alleles.)
- PI*M. The most common allele in all populations described to date. Some benign variants of the PI*M allele are designated M1, M2, M3, etc.
- PI*Z. The most common pathogenic allele , resulting in a quantitatively and functionally deficient AAT protein. Individuals homozygous for PI*Z (i.e., PI*ZZ) have severe alpha-1 antitrypsin deficiency (AATD).
- PI*S. A pathogenic allele resulting in a quantitatively and functionally deficient AAT. It is usually of clinical consequence only in the compound heterozygous state with another pathogenic allele (e.g., PI*SZ) and when the serum AAT level is <57 mg/dL.
- PI*F. A pathogenic allele that is distinctive because the resulting protein is functionally impaired in binding neutrophil elastase but quantitatively normal
- PI*I. An allele that is associated with mild quantitative deficiency
- Null alleles (sometimes designated PI*QO). Pathogenic alleles that result in either no mRNA product or no protein production
Molecular Genetic Testing Used in AATD
View in own window
See Table A. Genes and Databases for chromosome locus and protein.
See Molecular Genetics for information on variants detected in this gene .
Targeted analysis for pathogenic variants is typically specific for detecting the pathogenic alleles PI*Z and PI*S, which account for 95% of AATD [ McElvaney et al 1997 ].
Sequence analysis detects variants that are benign, likely benign , of uncertain significance , likely pathogenic , or pathogenic. Variants may include small intragenic deletions/insertions and missense , nonsense , and splice site variants; typically, exon or whole- gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here .
Gooptu et al [2014] , Greene et al [2016] , Hatipoğlu & Stoller [2016] , Matamala et al [2018] , Renoux et al [2018] , Strnad et al [2020]
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR , long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene -targeted microarray designed to detect single- exon deletions or duplications.
Rare exon and whole- gene deletions have been reported [ Takahashi & Crystal 1990 , Poller et al 1991 , Strnad et al 2020 ].
Detection of a Functionally Deficient AAT Protein Variant by Protease Inhibitor (PI) Typing
PI typing is performed by polyacrylamide gel isoelectric focusing (IEF) electrophoresis of serum in a gradient between pH 4 and 5. Note: IEF is no longer in common use in clinical practice.
- Electrophoretic AAT protein variants ( isoforms ) are designated by letters based on their migration pattern. For example, the normal AAT protein (designated M) migrates in the middle of the isoelectric field. The abnormal AAT deficiency protein (designated Z) migrates most slowly. Other variants have been given additional alphabetic designations; some rare variants have been named by place of origin of the proband .
- Because a range of AAT protein variants from normal to deficient can be observed in an IEF assay, a reference of 13 common and five rare AAT protein variants is used to identify the specific AAT protein [ Greene et al 2013 ].
- The limitations of IEF include inability to interpret an atypical electrophoretic pattern resulting from rare AAT protein variants and absence of AAT protein resulting from a SERPINA1 pathogenic null allele .
- IEF, the biochemical gold standard test for establishing the diagnosis of AATD, may be less costly than molecular genetic testing .
Though the optimal algorithm for laboratory testing is not well defined and recommendations in available guidelines differ [ Attaway et al 2019 ], the guidelines for the diagnosis and management of AATD by the American Thoracic Society / European Respiratory Society include recommended indications for genetic testing for AATD [ American Thoracic Society & European Respiratory Society 2003 ].
Clinical Indications for Genetic Testing
Adapted from Sandhaus et al [2016]
AATD = alpha-1 antitrypsin deficiency; COPD = chronic obstructive pulmonary disease; GPA = granulomatosis with polyangiitis
- Clinical Characteristics
Clinical Description
Alpha-1 antitrypsin deficiency (AATD) can present with hepatic dysfunction in individuals from infancy to adulthood and with obstructive lung disease and/or bronchiectasis, characteristically in individuals older than age 30 years. Phenotypic expression varies within and between families.
The severity of AATD depends on the genotype and resultant serum alpha-1 antitrypsin (AAT) level. Individuals homozygous for severe deficiency alleles (i.e., PI*ZZ) have low serum AAT levels, placing them at increased risk for chronic obstructive pulmonary disease (COPD) (see Table 4 ). Individuals with alleles associated with intrahepatic inclusions (e.g., Z, M malton, S iiyama ) are also at increased risk of developing liver disease.
Under-recognition of AATD often causes a long delay between first symptoms and initial diagnosis of AATD (i.e., 5-7 years) and many individuals report seeing multiple physicians before the diagnosis is first established. Diagnostic delay is associated with worsened clinical status at the time of initial diagnosis [ Tejwani et al 2019 ].
To date, approximately 5,000-10,000 individuals in the United States have been identified with a pathogenic variant in SERPINA1 [ American Thoracic Society & European Respiratory Society 2003 , Strnad et al 2020 ]. The following description of the phenotypic features associated with this condition is based on these reports.
Select Features of AATD
AAT = alpha-1 antitrypsin; AATD = alpha-1 antitrypsin deficiency; COPD = chronic obstructive pulmonary disease; GPA = granulomatosis with polyangiitis
Lung Disease
Adult-onset lung disease. Chronic obstructive pulmonary disease (COPD), specifically emphysema and/or chronic bronchitis, is the most common clinical manifestation of AATD. Bronchiectasis is also associated with AATD.
In adults, smoking is the major factor in accelerating the development of COPD. Although the natural history of AATD varies, depending in part on what has brought the individual to medical attention (e.g., lung symptoms, liver symptoms, asymptomatic relative of an affected individual), the onset of respiratory disease in smokers with AATD is characteristically between ages 40 and 50 years [ Tanash et al 2008 ]. Nonsmokers may have a normal life span, but can also develop lung and/or liver disease.
Individuals with severe AATD may manifest the usual signs and symptoms of obstructive lung disease, asthma, and chronic bronchitis (e.g., dyspnea, cough, wheezing, and sputum production) [ McElvaney et al 1997 ]. For example, in the National Heart, Lung, and Blood Institute Registry, of 1,129 participants with severe deficiency of AAT, 84% described dyspnea, 76% wheezed with an upper respiratory tract infection, and 50% reported cough and phlegm [ McElvaney et al 1997 , Eden et al 2003 ]. Of note, the prevalence of AATD in persons with asthma does not differ from that found in the general population [ Wencker et al 2002 , Miravitlles et al 2003 ].
Most individuals (~95%) with severe AATD have evidence of bronchiectasis on chest CT, with 27% demonstrating clinical symptoms of bronchiectasis [ Parr et al 2007 ].
- Chest CT shows loss of lung parenchyma and hyperlucency. In contrast to the usual pattern observed in centriacinar emphysema (emphysematous changes more pronounced in the lung apices than bases), the pattern observed in two thirds of individuals with AATD is that of more pronounced emphysematous changes in the bases than apices [ Parr et al 2004 ].
- Lung function tests show decreased expiratory airflow, increased lung volumes, and decreased diffusing capacity. Approximately 60% of individuals with AATD-associated emphysema demonstrate a component of reversible airflow obstruction, defined as a 200-mL and 12% increase in the post-bronchodilator FEV 1 and/or FVC.
Childhood-onset lung disease. Although reported, emphysema in children with AATD is extremely rare and may result from the coexistence of other unidentified genetic factors affecting the lung [ Cox & Talamo 1979 ].
Studies that followed newborns with severe AAT deficiency through age 32 years showed that most adults did not smoke and lacked physiologic and CT evidence of emphysema [ Mostafavi et al 2018 ]. Longer-term follow-up studies are not currently available. In most observational studies, the mean age of individuals with lung disease is in the fifth decade [ Seersholm et al 1997 , Alpha-1 Antitrypsin Deficiency Registry Study Group 1998 ].
Risk for lung disease in PI*MZ heterozygotes. Approximately 2%-3% of North Americans are PI*MZ heterozygotes. Nonsmoking PI*MZ heterozygotes are generally not considered to be at significantly increased risk for clinical emphysema [ Molloy et al 2014 ]. Specifically, population-based studies show no significant spirometric differences between matched PI*MZ and PI*MM cohorts [ Al Ashry & Strange 2017 ]. However, smoking PI*MZ heterozygotes are at increased risk for COPD [ Hersh et al 2004 , Sørheim et al 2010 , Molloy et al 2014 ]. Of note, slight abnormalities of lung function can be present without clinical symptoms. Alternatively, spirometry can miss at least 10% of individuals with a clinical diagnosis of COPD and emphysema on CT scan [ Smith et al 2014 , Lutchmedial et al 2015 ].
Risk for lung disease in persons with the PI*SZ genotype . Individuals who smoke and have the PI*SZ genotype with serum AAT levels below the protective threshold value have a slightly increased disease risk.
Relationship of AAT Protein Variants to Serum AAT Levels and Emphysema Risk in Adults
Adapted from Brantly et al [1991] , Stoller & Aboussouan [2005] , de Serres & Blanco [2012] , Bornhorst et al [2013]
AAT = alpha-1 antitrypsin; NA = North America
µmol/L
Note: An attempt to correlate serum AAT levels with protein variants in children showed trends similar to those seen in adults [ Donato et al 2012 ].
Liver Disease
Childhood-onset liver disease. The most common manifestation of AATD-associated liver disease is neonatal cholestasis: jaundice, with hyperbilirubinemia and raised serum aminotransferase levels in the early days and months of life.
Liver abnormalities develop in only a portion of children with AATD. In a study of 200,000 Swedish children who were followed up after newborn screening for AATD, 18% of those with the PI*ZZ genotype developed clinically recognized liver abnormalities and 2.4% developed liver cirrhosis with death in childhood [ Sveger 1976 , Sveger 1988 , Strnad et al 2020 ]. Liver damage may progress slowly [ Volpert et al 2000 ].
In a follow-up study of 44 children with AATD-associated liver disease initially manifesting as cirrhosis or portal hypertension, outcomes ranged from liver transplantation in two to relatively healthy lives up to 23 years after diagnosis in seven [ Migliazza et al 2000 ].
It is not known why only a small proportion of children with early hyperbilirubinemia have continued liver destruction leading to cirrhosis. The overall risk that an individual with the PI*ZZ genotype will develop severe liver disease in childhood is generally low (~2%); the risk is higher among sibs of a child with the PI*ZZ genotype and liver disease.
- When liver abnormalities in the proband are mild and resolve, the risk of liver disease in sibs with the PI*ZZ genotype is approximately 13%.
- When liver disease in the proband is severe, the risk for severe liver disease in sibs with the PI*ZZ genotype may be approximately 40% [ Cox 2004 ].
The PI*MZ and PI*SZ genotypes are not associated with an increased risk for childhood liver disease; however, on occasion, elevated levels of liver enzymes that resolve have been observed. In a study of 58 children with heterozygous genotypes showing signs of liver involvement during the first six months of life, almost all had normal values of liver enzymes at ages 12 months, five years, and ten years [ Pittschieler 2002 ].
Adult-onset liver disease. Liver disease in adults (manifesting as cirrhosis and fibrosis) may occur in the absence of a history of neonatal or childhood liver disease. Liver disease is more common in men than women.
The risk for liver disease at age 20-40 years is approximately 2% and at age 41-50 years approximately 4% [ Cox & Smyth 1983 ].
Autopsy studies suggest that the prevalence of liver disease may be as high as 40% in older individuals who have never smoked and do not have COPD [ Eriksson 1987 ]. Liver disease was subclinical at death in some of these individuals.
Hepatocellular carcinoma (HCC). The risk for HCC among individuals with AATD and the PI*ZZ genotype is several times that typically associated with liver cirrhosis. This increased risk has been attributed to failure of apoptosis of injured cells with retained Z protein, which sends a chronic regeneration signal to hepatocytes with a lesser load of retained Z protein [ Perlmutter 2006 ].
Liver pathology. AATD liver inclusions are visualized as bright pink globules of various sizes, using periodic acid-Schiff (PAS) stain following diastase treatment (PAS-D). The extent of inclusion formation varies considerably; the number and size of liver inclusions increases with age. Inclusions are not observed before age 12 weeks. Note: Liver biopsy, when indicated in the evaluation of individuals with liver disease, may show PAS positive diastase-resistant inclusion bodies which are suggestive of but not pathognomonic for AATD.
In infants with AATD, inclusions may be fine and granular and difficult to identify in percutaneous liver biopsy specimens. They are also observed in bile duct epithelium [ Cutz & Cox 1979 ].
Liver inclusions indicate the presence of at least one PI*Z allele ; histologic examination of the liver cannot confidently distinguish between PI*MZ heterozygotes and PI*ZZ homozygotes, although inclusions are generally more profuse in PI*ZZ homozygotes. Visualization of inclusions may be variable among PI*MZ heterozygotes.
Other Disease Associations
Panniculitis occurs in an estimated one in 1,000 individuals with AATD [ Alpha-1 Antitrypsin Deficiency Registry Study Group 1998 ]. Panniculitis characteristically presents as migratory, inflammatory, tender skin nodules which may ulcerate [ Stoller & Piliang 2008 ]. Sites of trauma (e.g., legs, lower abdomen) are most commonly affected. Presumably like emphysema in the lung, panniculitis in the skin is caused by unopposed proteolytic damage produced by the PI*Z allele .
Individuals with AATD appear to have increased susceptibility to C-ANCA-positive vasculitis (e.g., granulomatosis with polyangiitis [GPA], previously called Wegener granulomatosis) [ Hadzik-Blaszczyk et al 2018 ].
Genotype-Phenotype Correlations
The risk for lung disease associated with the following SERPINA1 genotypes is summarized in Table 4 .
PI*MM. This genotype is associated with a normal serum concentration of AAT and no increased risk of liver or lung disease.
PI*MZ. In general, nonsmoking individuals with this genotype are not considered to be at increased risk for lung disease; PI*MZ smokers and those with environmental exposures have increased risk of developing COPD [ Molloy et al 2014 , Al Ashry & Strange 2017 ].
PI*SS. This genotype does not appear to be associated with an increased risk for clinical disease [ Ferrarotti et al 2012 ]. The S allele is most common among individuals of Iberian descent.
PI*SZ. This genotype is not usually associated with a high risk for liver or lung disease; however, about 11% of individuals with the PI*SZ genotype have serum AAT levels below the protective threshold value (11 μM). Those individuals are at increased risk of developing emphysema with lower zone predominance, as well as chronic bronchitis, especially if they are smokers [ Green et al 2015 ].
PI*ZZ. Individuals with this genotype have a serum concentration of AAT that is approximately 10%-20% of normal (serum levels of 20-35 mg/dL) and are at high risk for both liver and lung disease. This genotype is present in 95% of affected individuals with clinical manifestations of AATD. Variable disease expressivity in individuals with the PI*ZZ genotype – not accounted for by the presence of known risk factors such as cigarette smoking – suggests the existence of other as-yet unidentified genetic disease modifiers.
PI*FF. While rare, the F allele is associated with AAT that is functionally impaired in binding neutrophil elastase but is quantitatively normal. Individuals with the PI*FF or PI*FZ genotype are deemed to be at increased risk of developing emphysema [ Sinden et al 2014 ].
PI* null -null (sometimes designated PI*QO). Individuals with this genotype have no measurable serum AAT secondary to complete lack of synthesis of AAT. Because protein does not accumulate in the liver, these individuals are not at increased risk of developing liver disease; however, they are at high risk of developing lung disease.
Nomenclature
In some publications, the term alpha-1-protease inhibitor is substituted for alpha-1 antitrypsin (AAT).
PI*M is used to describe normal alleles. Different normal alleles are given numeric designations (e.g., PI*M1, PI*M2).
AATD is one of the most common metabolic disorders in persons of northern European heritage, occurring in approximately one in 5,000-7,000 individuals in North America and one in 1,500-3,000 in Scandinavia. AATD also occurs (in lower frequencies) in all other racial subgroups worldwide [ Campbell 2000 , Miravitlles 2000 , de Serres & Blanco 2012 ].
- Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in SERPINA1 .
- Differential Diagnosis
Differential diagnoses include disorders causing chronic obstructive pulmonary disease (COPD), such as emphysema, chronic bronchitis, and bronchiectasis.
Bossé et al [2019] have described an autosomal dominant predisposition to emphysema in a single large French Canadian family that affects the protein, tyrosine phosphatase non-receptor, type 6 (PTPN6). As with severe deficiency of AAT, in this newly described condition there is near-complete penetrance for emphysema that is lower-lobe predominant and can be early onset (i.e., 4th-5th decade). Unlike alpha-1 antitrypsin deficiency (AATD), PTPN6 -type emphysema is inherited as an autosomal dominant condition.
See Table 5 for genetic disorders to consider in the differential diagnosis of AATD-related liver disease. The differential diagnosis of neonatal cholestasis also includes multiple metabolic diseases and other non-hereditary diseases including extrahepatic biliary atresia and gestational alloimmune liver disease (formerly known as neonatal hemochromatosis). Acquired disorders to consider include chronic viral hepatitis, alcoholic and non-alcoholic steatohepatitis, sclerosing cholangitis, and primary biliary cholangitis.
Genetic Disorders Associated with Liver Disease in the Differential Diagnosis of Alpha-1 Antitrypsin Deficiency
AATD = alpha-1 antitrypsin deficiency; AD = autosomal dominant ; ALT = alanine aminotransferase; AR = autosomal recessive ; AST = aspartate transaminase; GGT = gamma-glutamyl transferase; HFE = hemochromatosis; MOI = mode of inheritance
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with alpha-1 antitrypsin deficiency (AATD), the evaluations summarized in Table 6 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Recommended Evaluations Following Initial Diagnosis in Individuals with AATD
AATD = alpha-1 antitrypsin deficiency; MOI = mode of inheritance
Medical geneticist, certified genetic counselor, certified advanced genetic nurse
Treatment of Manifestations
Treatment of Manifestations in Individuals with AATD
AAT = alpha-1 antitrypsin; AATD = alpha-1 antitrypsin deficiency; COPD = chronic obstructive pulmonary disease; ICS = inhaled corticosteroids; LABA = long-acting beta agonists
Surveillance
Recommended Surveillance for Individuals with AATD
MRI = magnetic resonance imaging
Agents/Circumstances to Avoid
Avoid the following:
- Smoking (both active and passive)
- Occupational exposure (including exposure to environmental pollutants used in agriculture, mineral dust, gas, and fumes)
- Excessive use of alcohol
Evaluation of Relatives at Risk
The Alpha-1 Foundation-sponsored update of the ATS/ERS guidelines [ Sandhaus et al 2016 ] and the European Respiratory Society statement [ Miravitlles et al 2017 ] recommend evaluation of sibs, parents, and the children of an individual with severe AATD (see Table 2 ) in order to identify as early as possible those who would benefit from surveillance , institution of treatment, and preventive measures.
Extended pedigree analysis beyond first-degree relatives may be indicated in selected instances. For example, the presence of an AATD-associated condition (e.g., chronic obstructive pulmonary disease [COPD], liver disease, panniculitis) in a more distant family member and/or the finding that a parent of the proband has the PI*ZZ genotype would justify extensive family testing (i.e., of family members beyond parents, sibs, and offspring) [ Miravitlles et al 2017 ].
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Management of women with AATD during pregnancy should be guided by usual care principles, both for women without clinical disease and for those with liver disease. As noted, emphysema, especially in nonsmokers, would not commonly be expected during the usual childbearing age range.
Therapies Under Investigation
Many novel therapies for AATD are currently under investigation. Studies to slow the progression of lung disease include a variety of strategies: inhaled alpha-1 antitrypsin (AAT), liquid AAT, recombinant AAT, alternate dosing regimens of intravenous augmentation therapy (including double-dose strategies), an oral neutrophil elastase inhibitor, an orally available corrector molecule designed to restore secretion and acute phase reactivity, molecules to block polymer formation, and gene therapy using various viral vectors and delivery routes. Placement of valves endoscopically to improve lung function and functional status is also being studied. Examples of studies directed at the AATD-related liver disease include use of carbamazepine or sirolimus to increase autophagy and use of small interfering RNA to suppress aberrant AAT protein translation.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on these clinical studies.
- Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional . —ED.
Mode of Inheritance
Alpha-1 antitrypsin deficiency (AATD) is inherited in an autosomal codominant manner.
Risk to Family Members
Parents of a proband
- Either both parents of an individual with AATD are heterozygous for one SERPINA1 pathogenic variant (e.g., PI*MZ or PI*SZ) or, less frequently, a parent may be homozygous for the PI*Z allele (i.e., PI*ZZ).
- Molecular genetic testing is recommended for the parents of a proband to confirm their genetic status and to allow reliable recurrence risk assessment (see also Management, Evaluation of Relatives at Risk ).
- In general, nonsmoking heterozygotes are not considered to be at increased risk for lung disease; however, PI*MZ heterozygotes who have smoked are at increased risk for emphysema (see Genotype-Phenotype Correlations for additional information regarding risk of lung disease in heterozygotes).
Sibs of a proband
- If both parents are heterozygous (e.g., PI*MZ) for a pathogenic variant , each sib of an affected individual has a 25% chance of being affected (i.e., PI*ZZ), a 50% chance of being heterozygous (i.e., PI*MZ), and a 25% chance of inheriting neither of the pathogenic variants (i.e., PI*MM).
- If one parent is homozygous (i.e., PI*ZZ) for biallelic pathogenic variants and the other parent is heterozygous (e.g., PI*MZ) for a pathogenic variant , each sib has a 50% chance of being affected (i.e., PI*ZZ) and a 50% chance of being heterozygous (e.g., PI*MZ).
- Molecular genetic testing should be offered to all sibs in order to clarify their genetic status and identify as early as possible those who would benefit from surveillance , institution of treatment, and preventive measures.
Offspring of a proband
- Unless an individual with AATD has children with an affected individual or a heterozygote , offspring will be heterozygous for a pathogenic variant (e.g., PI*MZ).
- In populations with a high carrier frequency and/or a high rate of consanguinity , the reproductive partner of the proband may also have one or more SERPINA1 pathogenic variants. Thus, the risk to offspring is most accurately determined after (a) protease inhibitor (PI) typing by isoelectric focusing of serum or (b) SERPINA1 molecular genetic testing of the proband's reproductive partner.
Other family members. If the parents are heterozygous (e.g., PI*MZ) for a SERPINA1 pathogenic variant , each sib of the proband 's parents is at a 50% risk of being heterozygous for a pathogenic variant (e.g., PI*MZ).
Heterozygote Detection
Targeted molecular genetic testing for at-risk relatives requires prior identification of the SERPINA1 pathogenic variants in the family. If the pathogenic variants in the family have not been identified, heterozygote testing by protease inhibitor (PI) typing by isoelectric focusing of serum or SERPINA1 sequence analysis and deletion/duplication analysis are options.
Note: Measurement of serum AAT level is not reliable for determining carrier status because the range of serum AAT levels among most carriers may overlap the normal serum range [ Bornhorst et al 2013 ]. In addition, AAT is an acute-phase reactant and therefore serum AAT levels in a heterozygote may be elevated during periods of acute inflammation, thereby confounding the diagnosis of deficiency.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Risk to sibs of developing severe liver disease in infancy . Although the age of onset, severity, type of symptoms, and rate of progression of AATD cannot be predicted in sibs based on genotype , some estimates are available on the risk to sibs of developing severe liver disease in infancy [ Cox 2004 ].
- If the parents are heterozygotes (e.g., PI*MZ) but have not had a child with severe liver disease, the risk to offspring of having AATD (25%) AND severe liver disease in childhood (13.6%) is less than 1% (0.64%).
- If an affected individual died from severe liver disease in childhood, the risk to sibs of having AATD (25%) AND severe liver disease in childhood (40%) is 10%.
- If an affected individual did not have severe liver disease in childhood or if the liver disease resolved, the risk to sibs of having AATD (25%) AND liver disease (13%) is 3.3%.
Family planning. The optimal time for determination of genetic risk, clarification of genetic status, and discussion of the availability of prenatal/ preimplantation genetic testing is before pregnancy.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022] .
Prenatal Testing and Preimplantation Genetic Testing
Once the SERPINA1 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for AATD are possible.
Note: Prenatal testing is not useful in predicting age of onset, severity, type of symptoms, or rate of progression of the disorder. Fetal testing is not recommended in the American Thoracic Society/European Respiratory Society guidelines or in most other available guidelines [ Attaway et al 2019 ] because of the variable expressivity of disease and the possibility that individuals with severe deficiency of AAT can have a normal life span and escape disease, especially if they never smoke [ American Thoracic Society & European Respiratory Society 2003 ]. Because some children with AATD develop severe liver disease in the newborn period and some of these children have a poor outcome, prenatal diagnosis may be of interest to some at-risk couples who have previously had a child with severe liver disease (see Related Genetic Counseling Issues , Risk to sibs of developing severe liver disease in infancy ).
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing . While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here .
- Alpha-1 Advocacy Alliance 103 Rapidan Church Lane PO Box 202 Wolftown VA 22748 Phone: 866-367-2122 (toll-free); 540-948-6777 Fax: 540-948-6763 Email: [email protected] Alpha-1 Advocacy Alliance
- Alpha-1 Canada 13300 Tecumseh Road East, Suite 241 Tecumseh Ontario N8N 4R8 Canada Phone: 888-669-4583 (toll-free); 519-258-1444 Fax: 519-258-1614 Email: [email protected] www.alpha1canada.ca
- Alpha-1 Foundation 3300 Ponce de Leon Boulevard Coral Gables FL 33134 Phone: 877-228-7321; 305-567-9888 Fax: 305-567-1317 Email: [email protected] www.alpha-1foundation.org
- MedlinePlus Alpha-1 antitrypsin deficiency
- NCBI Genes and Disease Alpha -1-antitrypsin deficiency
- American Liver Foundation Phone: 800-465-4837 (HelpLine) www.liverfoundation.org
- Canadian Liver Foundation Canada Phone: 800-563-5483 Email: [email protected] www.liver.ca
- Childhood Liver Disease Research Network (ChiLDReN) Phone: 720-777-2598 Email: [email protected] www.childrennetwork.org
- Children's Liver Disease Foundation United Kingdom Phone: +44 (0) 121 212 3839 Email: [email protected] www.childliverdisease.org
- National Organization for Rare Disorders (NORD) Phone: 800-999-6673 Patient Assistance Programs
- Alpha-1 Canadian Registry Toronto Western Hospital 399 Bathurst Street 7th Floor, East Wing, Room 445 Toronto Ontario M5T 2S8 Canada Phone: 800-352-8186 (toll-free); 416-603-5020 Fax: 416-603-5020 Email: [email protected] www.alpha1canadianregistry.com
- Alpha-1 Research Registry Phone: 877-228-7321 ext 252 Email: [email protected] www.alpha1.org/investigators/resources/research-registry
- Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. — ED.
Alpha-1 Antitrypsin Deficiency: Genes and Databases
Data are compiled from the following standard references: gene from HGNC ; chromosome locus from OMIM ; protein from UniProt . For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click here .
OMIM Entries for Alpha-1 Antitrypsin Deficiency ( View All in OMIM )
Molecular Pathogenesis
SERPINA1 encodes alpha-1 antitrypsin (AAT), a glycoprotein member of the serum protease inhibitor (serpin) family. The molecule is composed of 418 amino acids; the first 24 are the signal peptide, while residues 25-418 encode the mature protein. AAT provides more than 90% of the protection against neutrophil elastase in the lower airways.
Mechanism of disease causation. The AAT disease mechanism can be either loss of function or gain of function.
- Lung disease. Alpha-1 antitrypsin deficiency (AATD) results in reduced inhibition of neutrophil elastase in the lung (which is increased in smokers), resulting in excessive destruction of the elastin in the alveolar walls. Thus, lung disease is considered to result from a loss-of-function mechanism.
- Liver disease. Abnormal AAT alleles (e.g., Z, M malton , S iiyama ) polymerize within hepatocytes [ Carrell & Lomas 2002 ], precluding secretion. Accumulation of abnormal AAT protein is associated with liver disease through a gain-of-function mechanism [ Kopito & Ron 2000 , Perlmutter 2002 ].
Protein variants, such as the PI*S variant, are more easily degraded. The PI*Z variant polymerizes within hepatocytes and alveolar macrophages where it was shown to be chemotactic for neutrophils. Thus, in addition to a loss-of-function mechanism, lung destruction may be fueled by an inflammatory reaction related to the polymers of Z protein variants in the lung [ McElvaney et al 1997 ].
SERPINA1 -specific laboratory technical considerations. Targeted testing for PI*Z, PI*S, PI*I, and PI*F is frequently performed. Differentiating targeted versus sequencing methods on a clinical report is important since targeted analysis detects about 95% of disease alleles.
Notable SERPINA1 Pathogenic Variants
AAT = alpha-1 antitrypsin; AATD = alpha-1 antitrypsin deficiency
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society ( varnomen .hgvs.org ). See Quick Reference for an explanation of nomenclature.
Variant designation that does not conform to current naming conventions; historical nomenclature does not include the signal sequence of the reference protein NP_000286 .3 , thereby decreasing the amino acid codon number by 24 amino acids for each variant.
- Chapter Notes
Author Notes
Research Support, Non-US Government
Research Support, US Government, PHS
Author History
Loutfi S Aboussouan, MD (2014-present) Diane W Cox, PhD, FCCMG, FRSC; University of Alberta (2005-2014) Felicitas L Lacbawan, MD; Cleveland Clinic (2014-2020) Kamilla Schlade-Bartusiak, PhD; University of Alberta (2005-2014) James K Stoller, MD, MS (2014-present) Vera Hupertz, MD (2020-present)
Revision History
- 1 June 2023 (aa) Revision: Table 4 : emphysema risk clarified to be lifetime emphysema risk
- 21 May 2020 (ha) Comprehensive update posted live
- 19 January 2017 (jks) Revision: clarification re serum levels of AAT in heterozygotes
- 1 May 2014 (me) Comprehensive update posted live
- 6 February 2008 (cd) Revision: sequence analysis available on a clinical basis
- 27 October 2006 (me) Review posted live
- 15 February 2005 (dc) Original submission
Published Guidelines / Consensus Statements
- American Thoracic Society, European Respiratory Society. American Thoracic Society / European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Available online . 2003. Accessed 5-23-23.
- Marciniuk DD, Hernandez P, Balter M, Bourbeau J, Chapman KR, Ford GT, Lauzon JL, Maltais F, O'Donnell DE, Goodridge D, Strange C, Cave AJ, Curren K, Muthuri S, et al. Alpha-1 antitrypsin deficiency targeted testing and augmentation therapy: a Canadian Thoracic Society clinical practice guideline. Available online . 2012. Accessed 5-23-23.
- Sandhaus RA, Turino G, Brantly M, Campos M, Cross C, Goodman K, Hogarth K, Knight S, Stocks J, Stoller JK, Strange C, Teckman J. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Available online . 2016. Accessed 5-23-23.
Literature Cited
- Al Ashry HS, Strange C. COPD in individuals with the PiMZ alpha-1 antitrypsin genotype. Eur Respir Rev. 2017; 26 :170068. [ PMC free article : PMC9488576 ] [ PubMed : 29070580 ]
- Alpha-1 Antitrypsin Deficiency Registry Study Group. Survival and FEV1 decline in individuals with severe deficiency of alpha-1 antitrypsin (Alpha-1 Antitrypsin Deficiency Registry Study Group). Am J Respir Crit Care Med. 1998; 158 :49–59. [ PubMed : 9655706 ]
- American Thoracic Society, European Respiratory Society. American Thoracic Society / European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha-1 antitrypsin deficiency. Am J Respir Crit Care Med. 2003; 168 :818–900. [ PubMed : 14522813 ]
- Attaway A, Majumdar U, Nowacki A, Sandhaus R, Stoller JK. An analysis of the degree of concordance among international guidelines regarding alpha-1 antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis. 2019; 14 :2089–101. [ PMC free article : PMC6734458 ] [ PubMed : 31564856 ]
- Bornhorst JA, Greene DN, Ashwood ER, Grenache DG. α1-Antitrypsin phenotypes and associated serum protein concentrations in a large clinical population. Chest. 2013; 143 :1000–8. [ PubMed : 23632999 ]
- Bossé Y, Lamontagne M, Gaudreault N, Racine C, Levesque MH, Smith BM, Auger D, Clemenceau A, Paré MÈ, Laviolette L, Tremblay V, Maranda B, Morissette MC, Maltais F. Early-onset emphysema in a large French-Canadian family: a genetic investigation. Lancet Respir Med. 2019; 7 :427–36. [ PubMed : 31000475 ]
- Brantly ML, Wittes JT, Vogelmeier CF, Hubbard RC, Fells GA, Crystal RG. Use of a highly purified alpha-1 antitrypsin standard to establish ranges for the common normal and deficient alpha-1 antitrypsin phenotypes. Chest. 1991; 100 :703–8. [ PubMed : 1889260 ]
- Campbell EJ. Alpha-1 antitrypsin deficiency: incidence and detection program. Respir Med. 2000;94 Suppl C:18-21. [ PubMed : 10954251 ]
- Carrell RW, Lomas DA. Alpha-1 antitrypsin deficiency - a model for conformational diseases. N Engl J Med. 2002; 346 :45–53. [ PubMed : 11778003 ]
- Corda L, Bertella E, La Piana GE, Boni E, Redolfi S, Tantucci C. Inhaled corticosteroids as additional treatment in alpha-1-antitrypsin-deficiency-related COPD. Respiration. 2008; 76 :61–8. [ PubMed : 18319586 ]
- Cox DW. Prenatal diagnosis for alpha-1 antitrypsin deficiency. Prenat Diagn. 2004; 24 :468–70. [ PubMed : 15229848 ]
- Cox DW, Johnson AM, Fagerhol M. Report of nomenclature meeting for alpha-1 antitrypsin. Hum Genet. 1980; 53 :429–33. [ PubMed : 6102963 ]
- Cox DW, Smyth S. Risk for liver disease in adults with alpha-1 antitrypsin deficiency. Am J Med. 1983; 74 :221–7. [ PubMed : 6600583 ]
- Cox DW, Talamo RC. Genetic aspects of pediatric lung disease. Pediatr Clin North Am. 1979; 26 :467–80. [ PubMed : 315047 ]
- Cutz E, Cox DW. Alpha-1 antitrypsin deficiency: the spectrum of pathology and pathophysiology. Perspect Pediatr Pathol. 1979; 5 :1–39. [ PubMed : 231756 ]
- de Serres FJ, Blanco I. Prevalence of α1-antitrypsin deficiency alleles PI*Sand PI*Z worldwide and effective screening for each of the five phenotypic classes PI*MS, PI*MZ, PI*SS, PI*SZ, and PI*ZZ: a comprehensive review. Ther Adv Respir Dis. 2012; 6 :277–95. [ PubMed : 22933512 ]
- Donato LJ, Jenkins SM, Smith C, Katzmann JA, Snyder MR. Reference and interpretive ranges for α(1)-antitrypsin quantitation by phenotype in adult and pediatric populations. Am J Clin Pathol. 2012; 138 :398–405. [ PubMed : 22912357 ]
- Eden E, Hammel J, Rouhani FN, Brantly ML, Barker AF, Buist AS, Fallat RJ, Stoller JK, Crystal RG, Turino GM. Asthma features in severe alpha-1 antitrypsin deficiency: experience of the National Heart, Lung, and Blood Institute Registry. Chest. 2003; 123 :765–71. [ PubMed : 12628876 ]
- Eriksson S. Alpha-1 antitrypsin deficiency and liver cirrhosis in adults. An analysis of 35 Swedish autopsied cases. Acta Med Scand. 1987; 221 :461–7. [ PubMed : 3496734 ]
- Ferrarotti I, Thun GA, Zorzetto M, Ottaviani S, Imboden M, Schindler C, von Eckardstein A, Rohrer L, Rochat T, Russi EW, Probst-Hensch NM, Luisetti M. Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax. 2012; 67 :669–74. [ PubMed : 22426792 ]
- Gooptu B, Dickens JA, Lomas DA. The molecular and cellular pathology of α 1 -antitrypsin deficiency. Trends Mol Med. 2014; 20 :116–27. [ PubMed : 24374162 ]
- Graham A, Kalsheker NA, Newton CA, et al. Molecular characterization of three alpha-1 antitrypsin deficiency variants: proteinase inhibitor (Pi) null Cardiff (Asp 256 to Val); Mi M malton (Phe 51 to deletion) and Pi I (Arg 39 to Cys). Hum Genet. 1989; 84 :55–8. [ PubMed : 2606478 ]
- Green CE, Vayalapra S, Hampson JA, Mukherjee D, Stockley RA, Turner AM. PiSZ alpha-1 antitrypsin deficiency (AATD): pulmonary phenotype and prognosis relative to PiZZ AATD and PiMM COPD. Thorax. 2015; 70 :939–45. [ PubMed : 26141072 ]
- Greene CM, Marciniak S, Teckman J, Ferrarotti I, Brantly M, Lomas D, Stoller JK, McElvaney N. α-1 antitrypsin deficiency. Nat Rev Dis Primers. 2016; 2 :16051. [ PubMed : 27465791 ]
- Greene DN, Elliott-Jelf MC, Straseski JA, Grenache DG. Facilitating the laboratory diagnosis of α1-antitrypsin deficiency. Am J Clin Pathol. 2013; 139 :184–91. [ PubMed : 23355203 ]
- Hadzik-Blaszczyk M, Zdral A, Zielonka TM, Rozy A, Krupa R, Falkowski A, Wardyn KA, Chorostowska-Wynimko J. Zycinska K l. SERPINA1 Gene variants in granulomatosis with polyangiitis. Adv Exp Med Biol. 2018; 1070 :9–18. [ PubMed : 29460271 ]
- Hatipoğlu U, Stoller JK. α1-Antitrypsin Deficiency. Clin Chest Med. 2016; 37 :487–504. [ PubMed : 27514595 ]
- Hersh CP, Dahl M, Ly NP, Berkey CS, Nordestgaard BG, Silverman EK. Chronic obstructive pulmonary disease in alpha-1 antitrypsin PI MZ heterozygotes: a meta-analysis. Thorax. 2004; 59 :843–9. [ PMC free article : PMC1746834 ] [ PubMed : 15454649 ]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022; 13 :389–97. [ PMC free article : PMC9314484 ] [ PubMed : 35834113 ]
- Kleinerova J, Ging P, Rutherford C, Lawrie I, Winward S, Eaton D, Redmond KC, Egan JJ. The withdrawal of replacement therapy and outcomes in alpha-1 antitrypsin deficiency lung transplant recipients. Eur Respir J. 2019; 53 :1900055. [ PubMed : 30819816 ]
- Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000; 2 :E207–9. [ PubMed : 11056553 ]
- Lutchmedial SM, Creed WG, Moore AJ, Walsh RR, Gentchos GE, Kaminsky DA. How common is airflow limitation in patients with emphysema on CT scan of the chest? Chest. 2015; 148 :176–84. [ PMC free article : PMC4493873 ] [ PubMed : 25539080 ]
- Mahr AD, Edberg JC, Stone JH, Hoffman GS, St Clair EW, Specks U, Dellaripa PF, Seo P, Spiera RF, Rouhani FN, Brantly ML, Merkel PA. Alpha-1 antitrypsin deficiency-related alleles Z and S and the risk of Wegenerʼs granulomatosis. Arthritis Rheum. 2010; 62 :3760–7. [ PMC free article : PMC3123032 ] [ PubMed : 20827781 ]
- Matamala N, Lara B, Gomez-Mariano G, Martínez S, Retana D, Fernandez T, Silvestre RA, Belmonte I, Rodriguez-Frias F, Vilar M, Sáez R, Iturbe I, Castillo S, Molina-Molina M, Texido A, Tirado-Conde G, Lopez-Campos JL, Posada M, Blanco I, Janciauskiene S, Martinez-Delgado B. Characterization of novel missense variants of SERPINA1 gene causing alpha-1 antitrypsin deficiency. Am J Respir Cell Mol Biol. 2018; 58 :706–16. [ PubMed : 29232161 ]
- McElvaney NG, Stoller JK, Buist AS, Prakash UB, Brantly ML, Schluchter MD, Crystal RD. Baseline characteristics of enrollees in the National Heart, Lung and Blood Institute Registry of alpha-1 antitrypsin deficiency. Alpha-1 Antitrypsin Deficiency Registry Study Group. Chest. 1997; 111 :394–403. [ PubMed : 9041988 ]
- McElvaney OJ, Carroll TP, Franciosi AN, Sweeney J, Hobbs BD, Kowlessar V, Gunaratnam C, Reeves EP, McElvaney NG. Consequences of abrupt cessation of alpha(1)-antitrypsin replacement therapy. N Engl J Med. 2020; 382 :1478–80. [ PubMed : 32268034 ]
- Migliazza L, Lopez Santamaria M, Murcia J, Gamez M, Clavijo J, Camarena C, Hierro L, Frauca E, de la Vega A, Diaz M, Jara P, Tovar JA. Long-term survival expectancy after liver transplantation in children. J Pediatr Surg. 2000; 35 :5–7. [ PubMed : 10646764 ]
- Miravitlles M. Alpha-1 antitrypsin deficiency: epidemiology and prevalence. Respir Med. 2000; 94 Suppl C:S12-5. [ PubMed : 10954249 ]
- Miravitlles M, Dirksen A, Ferrarotti I, Koblizek V, Lange P, Mahadeva R, McElvaney NG, Parr D, Piitulainen E, Roche N, Stolk J, Thabut G, Turner A, Vogelmeier C, Stockley RA. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α(1)-antitrypsin deficiency. Eur Respir J. 2017; 50 :1700610. [ PubMed : 29191952 ]
- Miravitlles M, Vila S, Jardi R, de la Roza C, Rodriguez-Frias F, Vidal R. Emphysema due to alpha-1 antitrypsin deficiency: familial study of the YBARCELONA variant. Chest. 2003; 124 :404–6. [ PubMed : 12853554 ]
- Molloy K, Hersh CP, Morris VB, Carroll TP, O'Connor CA, Lasky-Su JA, Greene CM, O'Neill SJ, Silverman EK, McElvaney NG. Clarification of the risk of chronic obstructive pulmonary disease in alpha-1 antitrypsin deficiency PiMZ heterozygotes. Am J Respir Crit Care Med. 2014; 189 :419–27. [ PMC free article : PMC5955067 ] [ PubMed : 24428606 ]
- Mostafavi B, Diaz S, Piitulainen E, Stoel BC, Wollmer P, Tanash HA. Lung function and CT lung densitometry in 37-39 year old individuals with alpha-1 antitrypsin deficiency. Int J Chron Obstruct Pulmon Dis. 2018; 13 :3689–98. [ PMC free article : PMC6231508 ] [ PubMed : 30510411 ]
- Parr DG, Guest PG, Reynolds JH, Dowson LJ, Stockley RA. Prevalence and impact of bronchiectasis in alpha-1antitrypsin deficiency. Am J Respir Crit Care Med. 2007; 176 :1215–21. [ PubMed : 17872489 ]
- Parr DG, Stoel BC, Stolk J, Stockley RA. Pattern of emphysema distribution in alpha-1 antitrypsin deficiency influences lung function impairment. Am J Respir Crit Care Med. 2004; 170 :1172–8. [ PubMed : 15306534 ]
- Perlmutter DH. Liver injury in alpha-1 antitrypsin deficiency: an aggregated protein induces mitochondrial injury. J Clin Invest. 2002; 110 :1579–83. [ PMC free article : PMC151639 ] [ PubMed : 12464659 ]
- Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1 antitrypsin deficiency. Pediatr Res. 2006; 60 :233–8. [ PubMed : 16864711 ]
- Pittschieler K. Liver involvement in alpha-1 antitrypsin-deficient phenotypes PiSZ and PiMZ. Acta Paediatr. 2002; 91 :239–40. [ PubMed : 11952016 ]
- Poller W, Faber JP, Weidinger S, Olek K. DNA polymorphisms associated with a new alpha 1-antitrypsin PIQ0 variant (PIQ0riedenburg). Hum Genet. 1991; 86 :522–4. [ PubMed : 1673114 ]
- Renoux C, Odou MF, Tosato G, Teoli J, Abbou N, Lombard C, Zerimech F, Porchet N, Chapuis Cellier C, Balduyck M, Joly P. Description of 22 new alpha-1 antitrypsin genetic variants. Orphanet J Rare Dis. 2018; 13 :161. [ PMC free article : PMC6142351 ] [ PubMed : 30223862 ]
- Sandhaus RA, Turino G, Brantly ML, Campos M, Cross CE, Goodman K, Hogarth DK, Knight SL, Stocks JM, Stoller JK, Strange C, Teckman J. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis. 2016; 3 :668–82. [ PMC free article : PMC5556762 ] [ PubMed : 28848891 ]
- Seersholm N, Wencker M, Banik N, Viskum K, Dirksen A, Kok-Jensen A, Konietzko N. Does alpha-1 antitrypsin augmentation therapy slow the annual decline in FEV1 in patients with severe hereditary alpha-1 antitrypsin deficiency? Wissenschaftliche Arbeitsgemeinschaft zur Therapie von Lungenerkrankungen (WATL) Alpha-1 AT study group. Eur Respir J. 1997; 10 :2260–3. [ PubMed : 9387950 ]
- Smith BM, Austin JH, Newell JD Jr, D'Souza BM, Rozenshtein A, Hoffman EA, Ahmed F, Barr RG. Pulmonary emphysema subtypes on computed tomography: the MESA COPD study. Am J Med. 2014; 127 :94.e7–23. [ PMC free article : PMC3882898 ] [ PubMed : 24384106 ]
- Sinden NJ, Koura F, Stockley RA. The significance of the F variant of alpha-1 antitrypsin and unique case report of a PiFF homozygote. BMC Pulm Med. 2014; 14 :132–9. [ PMC free article : PMC4131482 ] [ PubMed : 25098359 ]
- Sørheim IC, Bakke P, Gulsvik A, Pillai SG, Johannessen A, Gaarder PI, Campbell EJ, Agustí A, Calverley PM, Donner CF, Make BJ, Rennard SI, Vestbo J, Wouters EF, Paré PD, Levy RD, Coxson HO, Lomas DA, Hersh CP, Silverman EK. α 1 -antitrypsin protease inhibitor MZ heterozygosity is associated with airflow obstruction in two large cohorts. Chest. 2010; 138 :1125–32. [ PMC free article : PMC2972629 ] [ PubMed : 20595457 ]
- Spratt JR, Brown RZ, Rudser K, Goswami U, Hertz MI, Patil J, Cich I, Shumway SJ, Loor G. Greater survival despite increased complication rates following lung transplant for alpha-1-antitrypsin deficiency compared to chronic obstructive pulmonary disease. J Thorac Dis. 2019; 11 :1130–44. [ PMC free article : PMC6531741 ] [ PubMed : 31179055 ]
- Stoller JK, Aboussouan LS. Alpha-1 antitrypsin deficiency. Lancet. 2005; 365 :2225–36. [ PubMed : 15978931 ]
- Stoller JK, Piliang M. Panniculitis in alpha-1 antitrypsin deficiency: a review. Clin Pulm Med. 2008; 15 :113–7.
- Strnad P, McElvaney NG, Lomas DA. Alpha-1 antitrypsin deficiency. New Engl J Med. 2020; 382 :1443–55. [ PubMed : 32268028 ]
- Sveger T. Liver disease in alpha-1 antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. 1976; 294 :1316–21. [ PubMed : 1083485 ]
- Sveger T. The natural history of liver disease in alpha-1 antitrypsin deficient children. Acta Paediatr Scand. 1988; 77 :847–51. [ PubMed : 2905108 ]
- Takahashi H, Crystal RG. Alpha 1-antitrypsin Null(isola di procida): an alpha 1-antitrypsin deficiency allele caused by deletion of all alpha 1-antitrypsin coding exons. Am J Hum Genet. 1990; 47 :403–13. [ PMC free article : PMC1683852 ] [ PubMed : 1975477 ]
- Tanash HA, Nilsson PM, Nilsson JA, Piitulainen E. Clinical course and prognosis of never-smokers with severe alpha-1 antitrypsin deficiency (PiZZ). Thorax. 2008; 63 :1091–5. [ PubMed : 18682522 ]
- Tejwani V, Nowacki A, Fye E, Sanders C, Stoller JK. The impact of delayed diagnosis of alpha-1 antitrypsin deficiency: the association between diagnostic delay and worsened clinical status. Respir Care. 2019; 64 :915–22. [ PubMed : 30914495 ]
- Volpert D, Molleston JP, Perlmutter DH. Alpha-1 antitrypsin deficiency-associated liver disease progresses slowly in some children. J Pediatr Gastroenterol Nutr. 2000; 31 :258–63. [ PubMed : 10997369 ]
- Wencker M, Marx A, Konietzko N, Schaefer B, Campbell EJ. Screening for alpha-1 Pi deficiency in patients with lung diseases. Eur Respir J. 2002; 20 :319–24. [ PubMed : 12212962 ]
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source ( http://www.genereviews.org/ ) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer . No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer .
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda .
- Cite this Page Stoller JK, Hupertz V, Aboussouan LS. Alpha-1 Antitrypsin Deficiency. 2006 Oct 27 [Updated 2023 Jun 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
- PDF version of this page (597K)
- Disable Glossary Links
In this GeneReview
Bulk download.
- Bulk download GeneReviews data from FTP
GeneReviews Links
- GeneReviews Glossary
- Resource Materials NEW FEATURE
- New in GeneReviews
- Author List
- For Current/Prospective Authors
- GeneReviews Personnel
- Download/Link to GeneReviews
Tests in GTR by Gene
Related information.
- MedGen Related information in MedGen
- OMIM Related OMIM records
- PMC PubMed Central citations
- PubMed Links to PubMed
- Gene Locus Links
Similar articles in PubMed
- Review Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. [Orphanet J Rare Dis. 2008] Review Hereditary alpha-1-antitrypsin deficiency and its clinical consequences. Fregonese L, Stolk J. Orphanet J Rare Dis. 2008 Jun 19; 3:16. Epub 2008 Jun 19.
- Alpha-1 Antitrypsin Augmentation and the Liver Phenotype of Adults With Alpha-1 Antitrypsin Deficiency (Genotype Pi∗ZZ). [Clin Gastroenterol Hepatol. 2024] Alpha-1 Antitrypsin Augmentation and the Liver Phenotype of Adults With Alpha-1 Antitrypsin Deficiency (Genotype Pi∗ZZ). Fromme M, Hamesch K, Schneider CV, Mandorfer M, Pons M, Thorhauge KH, Pereira V, Sperl J, Frankova S, Reichert MC, et al. Clin Gastroenterol Hepatol. 2024 Feb; 22(2):283-294.e5. Epub 2023 Sep 15.
- Review Non-Invasive Assessment and Management of Liver Involvement in Adults With Alpha-1 Antitrypsin Deficiency. [Chronic Obstr Pulm Dis. 2020] Review Non-Invasive Assessment and Management of Liver Involvement in Adults With Alpha-1 Antitrypsin Deficiency. Hamesch K, Strnad P. Chronic Obstr Pulm Dis. 2020 Jul; 7(3):260-271.
- Clinical presentations of four patients with rare Alpha 1 Antitrypsin variants identified in a single US center. [Respir Med Case Rep. 2021] Clinical presentations of four patients with rare Alpha 1 Antitrypsin variants identified in a single US center. Kueppers F. Respir Med Case Rep. 2021; 32:101345. Epub 2021 Jan 20.
- Liver Phenotypes of European Adults Heterozygous or Homozygous for Pi∗Z Variant of AAT (Pi∗MZ vs Pi∗ZZ genotype) and Noncarriers. [Gastroenterology. 2020] Liver Phenotypes of European Adults Heterozygous or Homozygous for Pi∗Z Variant of AAT (Pi∗MZ vs Pi∗ZZ genotype) and Noncarriers. Schneider CV, Hamesch K, Gross A, Mandorfer M, Moeller LS, Pereira V, Pons M, Kuca P, Reichert MC, Benini F, et al. Gastroenterology. 2020 Aug; 159(2):534-548.e11. Epub 2020 May 4.
Recent Activity
- Alpha-1 Antitrypsin Deficiency - GeneReviews® Alpha-1 Antitrypsin Deficiency - GeneReviews®
Your browsing activity is empty.
Activity recording is turned off.
Turn recording back on
Connect with NLM
National Library of Medicine 8600 Rockville Pike Bethesda, MD 20894
Web Policies FOIA HHS Vulnerability Disclosure
Help Accessibility Careers
Alpha-1 Antitrypsin Deficiency
- Diagnosis |
- Treatment |
Alpha-1 antitrypsin deficiency is a hereditary disorder in which a lack or low level of the enzyme alpha-1 antitrypsin damages the lungs and liver.
Alpha-1 antitrypsin deficiency is caused by an inherited gene mutation.
Infants may develop jaundice and liver damage.
Cirrhosis can develop during childhood.
Adults commonly develop emphysema, with shortness of breath, wheezing, and coughing, and some adults develop cirrhosis.
Tests that measure the amount of the enzyme in the blood and that detect the gene mutations are used for diagnosis.
People with emphysema take medications to improve breathing and sometimes receive infusions of alpha-1 antitrypsin by vein.
Some people need lung or liver transplants.
Alpha-1 antitrypsin is an enzyme produced by the liver that inhibits the action of other enzymes called proteases. Proteases break down proteins as part of normal tissue repair. Alpha-1 antitrypsin protects the lungs from the damaging effects of proteases.
Alpha-1 antitrypsin deficiency results from an inherited mutation in the gene that controls production and release of the enzyme. There are many subtypes of alpha-1 antitrypsin deficiency, but in all, levels of active enzyme in the blood are insufficient, the enzyme is structurally abnormal (and thus functions poorly), or both. People of Northern European ancestry are affected more often than people of African, Asian, or Hispanic ancestry.
The most common problems caused by the deficiency are
Liver damage
Emphysema (a form of chronic obstructive pulmonary disease , or COPD)
If the enzyme is structurally abnormal, it may clump in the liver, causing the liver to malfunction. In some people, liver malfunction leads to cirrhosis and to an increased risk of liver cancer.
The low levels of alpha-1 antitrypsin allow proteases to damage the lungs, resulting in emphysema . Emphysema is more common (and worse) in people who smoke. Emphysema in people who do not smoke can be caused by alpha-1 antitrypsin deficiency.
Disorders of other organs sometimes occur. These disorders include inflammation of fat under the skin ( panniculitis ), life-threatening bleeding, aneurysms , ulcerative colitis , vasculitis , and kidney disease.
Symptoms of Alpha-1 Antitrypsin Deficiency
Symptoms may first appear during infancy, childhood, or adulthood. About 10 to 20% of affected people have symptoms during infancy. Affected infants develop yellowing of the skin and the whites of the eyes ( jaundice ) and an enlarged liver during the first week of life. Jaundice disappears at about age 2 to 4 months. However, about 20% of these infants later develop cirrhosis, and some die before reaching adulthood.
Adults commonly develop emphysema, with progressively increasing shortness of breath, difficulty breathing, coughing, and wheezing. Emphysema rarely develops before age 25 years. It develops earlier and is more severe in people who smoke than in people who do not. The severity of symptoms also varies depending on the form of the deficiency, other disorders people have, environmental exposure to lung irritants, and other factors. If people have never smoked, their symptoms tend to be moderate, and most have a normal life expectancy.
Even if they did not have liver problems during infancy, about 10% of adults develop cirrhosis, which may eventually lead to liver cancer.
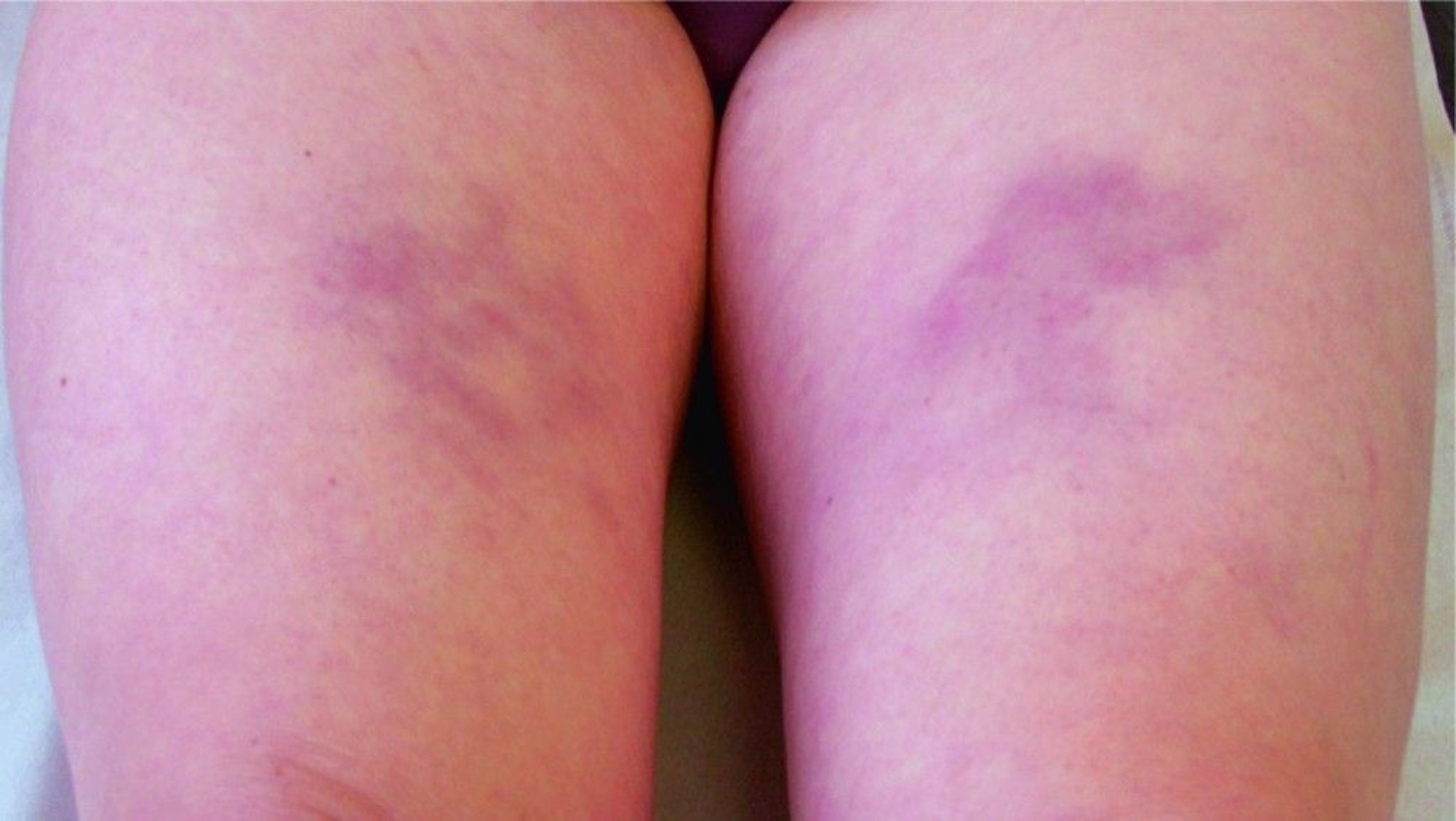
© Springer Science+Business Media
People with panniculitis have painful, tender bumps or discolored patches on the lower abdomen, buttocks, and thighs. The bumps may feel hard to the touch.
Diagnosis of Alpha-1 Antitrypsin Deficiency
Alpha-1 antitrypsin deficiency is suspected in the following:
Infants who have typical symptoms
People who smoke who develop emphysema before age 45 years
People who do not smoke who develop emphysema at any age
People with an unexplained liver disorder
People who develop panniculitis
People with a family history of emphysema or unexplained cirrhosis
People with a family history of alpha-1 antitrypsin deficiency
Because the deficiency is inherited, doctors usually ask whether any family members have had emphysema or cirrhosis with no known cause.
The deficiency is usually confirmed by genetic testing, which also can determine the specific form of the deficiency. Doctors also usually do blood tests to measure the level of alpha-1 antitrypsin.
Treatment of Alpha-1 Antitrypsin Deficiency
People who smoke are advised to stop
Alpha-1 antitrypsin may be given by vein to replace the deficient enzyme. It is collected from a group of donors and screened for bloodborne disorders. Thus, it is expensive and is most beneficial to people who have only moderate symptoms due to emphysema and who do not smoke. This treatment is thought to prevent further damage but does not reverse damage already done.
If people are younger than 60 years and have severe symptoms, lung transplantation may be done. A few medical centers sometimes do transplantations in highly selected people as old as 70 years. A few medical centers also sometimes do lung volume reduction surgery .
Taking alpha-1 antitrypsin does not treat or prevent liver damage because liver damage is caused by production of an abnormal enzyme, not by enzyme deficiency. If the liver is severely damaged, liver transplantation may be done. The transplanted liver does not become damaged because the alpha-1 antitrypsin it produces is normal and thus does not accumulate in the liver.
Panniculitis

Copyright © 2024 Merck & Co., Inc., Rahway, NJ, USA and its affiliates. All rights reserved.
- Cookie Preferences

An official website of the United States government
Here’s how you know
Official websites use .gov A .gov website belongs to an official government organization in the United States.
Secure .gov websites use HTTPS A lock ( Lock Locked padlock icon ) or https:// means you’ve safely connected to the .gov website. Share sensitive information only on official, secure websites.

- Health Topics
- Drugs & Supplements
- Medical Tests
- Medical Encyclopedia
- About MedlinePlus
- Customer Support
Alpha-1 Antitrypsin Testing
What is alpha-1 antitrypsin (aat) testing.
Alpha-1 antitrypsin (AAT) testing uses a sample of blood or a cheek swab to diagnose a condition called alpha-1 antitrypsin deficiency (AAT deficiency). This condition is sometimes known as "alpha-." or AATD. If you have AAT deficiency, your body doesn't make enough AAT.
AAT is made by your liver. It helps protect your lungs from inflammation and irritating substances you might breathe in, such as smoke. If your liver doesn't make enough AAT, your lungs may be more easily damaged from smoking , pollution , or dust from the environment. This can lead to a serious lung condition called chronic obstructive pulmonary disease ( COPD ). AAT deficiency may also cause a liver disease called cirrhosis . This is more common in children who have AAT deficiency.
AAT deficiency is a genetic disorder . That means it's caused by changes in your genes, which may also be called gene variants or mutations.
Genes carry information that controls what you look like and how your body works. AAT deficiency is caused by changes in the SERPINA1 gene , which carries instructions for making the AAT protein. These gene changes are inherited from your parents, so AAT deficiency tends to run in families.
- If you have two mutated copies of the gene , it means you have a condition called AAT deficiency. People with this disorder have a higher risk of getting lung disease or liver damage before the age of 45.
- If you have one mutated copy of the gene , you are a carrier of AAT deficiency. In these cases, this means you are at slightly higher risk of developing lung disease, especially if you other risk factors, such as being a smoker. You could pass the mutated gene on to your children.
There are a few gene changes that cause AAT deficiency. These gene changes can:
- Decrease the amount of AAT protein your liver makes.
- Prevent your liver from making any AAT.
- Affect the shape of the AAT protein so that it can't move out of your liver to protect your lungs. Over time, AAT builds up in your liver and causes damage.
A genetic test can help you find out whether you have the gene change that increases your risk for lung and liver disease.
There are three types of testing to help diagnose AAT deficiency:
- If your AAT levels are abnormally low, a genetic test (either a genotype test or a phenotype test) is needed to confirm a diagnosis of AAT deficiency.
- The genotype test looks for the more common types of gene changes that can cause AAT deficiency.
- The phenotype test checks for changes in the AAT protein that change how it would normally work.
Other names: A1AT, AAT, alpha-1-antiprotease deficiency, α1-antitrypsin, serum AAT test, AAT phenotyping, AAT genotyping, AAT deficiency test, AAT DNA sequencing test, AAT isoelectric focusing test, A1AT test, AATD test, alpha-1 protease inhibitor deficiency test
What is it used for?
There are three main uses for AAT testing:
- To diagnose or rule out AAT deficiency in people who have symptoms of lung and/or liver disease that may be caused by AAT deficiency
- To screen people who don't have symptoms, but have family members with AAT deficiency
- To guide treatment choices for lung disease that may be caused by AAT deficiency
Why do I need AAT testing?
Diagnostic AAT testing may be recommended if you have the signs and symptoms of AAT deficiency or if you have conditions that could be caused by AAT deficiency.
Signs and symptoms of AAT deficiency may include:
- Shortness of breath after exercise
- Chronic cough with phlegm (mucus)
- Faster-than-normal heartbeat when you stand up
- Vision problems
Conditions that could be caused by AAT deficiency can include:
- COPD (chronic obstructive pulmonary disease)
- Repeated respiratory infections, such as colds and bronchitis
- Asthma that doesn't respond well to treatment
- Panniculitis, a skin condition that causes hardened skin with painful lumps or patches
- Liver disease without a known cause
- Jaundice (yellowing of the skin and eyes)
You may also get this test if you have a family history of AAT deficiency, emphysema , or unexplained cirrhosis.
AAT deficiency in babies often affects the liver. Your baby may need AAT testing if he or she has signs of liver disease such as jaundice or abnormal liver enzyme tests .
What happens during AAT testing?
AAT blood testing may involve a blood test with blood from your arm or a finger prick. Some genetics tests involve a cheek swab.
For a blood test from your arm , a health care professional will take the blood sample from a vein in your arm, using a small needle. After the needle is inserted, a small amount of blood will be collected into a test tube or vial. You may feel a little sting when the needle goes in or out. This usually takes less than five minutes.
For a finger prick test , a health care professional will prick your fingertip to obtain a small amount of blood.
For a cheek swab , a health care professional will wipe the inside of your cheek with a small tool to remove some cells. You may have the option of doing it yourself.
Will I need to do anything to prepare for the test?
You don't need any special preparations for an AAT test.
Are there any risks to the test?
There is very little physical risk to a blood test. If the blood was taken from your arm, you may have slight pain or bruising at the spot where the needle was put in, but most symptoms go away quickly.
There are no risks to having a cheek swab.
What do the results mean?
If your AAT blood levels are low, it may mean you have AAT deficiency. To confirm the diagnosis, you'll need a genotype or phenotype test.
If that testing shows that you have AAT deficiency, your provider may recommend additional testing to evaluate your lung and liver function.
There is no cure for AAT deficiency. But treatments can help prevent your condition from getting worse. Your exact treatment will depend on your symptoms.
You can also take steps to stay as healthy as possible, including:
- Not smoking. If you are a smoker, quit smoking . If you don't smoke, don't start. Smoking is the leading risk factor for life-threatening lung disease in people with AAT deficiency.
- Not drinking alcohol . Alcohol can damage your liver.
- Following a healthy diet.
- Getting regular exercise .
- Seeing your provider regularly.
- Taking medicines as prescribed by your provider.
If you have questions about your results, talk to your provider.
Learn more about laboratory tests, reference ranges, and understanding results .
Is there anything else I need to know about AAT testing?
If you're thinking about having AAT testing or have received abnormal results, it may be helpful to speak with a genetic counselor . A genetic counselor is a specially trained professional in genetics and genetic testing. A counselor can help you understand the risks and benefits of testing for you and your family. After testing, a counselor can help you understand your results. If you have AAT deficiency, the counselor can provide information on the condition, including your risk of passing the disease to your children.
- Alpha-1 Foundation [Internet]. Coral Gables (FL): Alpha-1 Foundation. c2023. Testing for Alpha-1; [cited 2023 Jun 14]; [about 3 screens]. Available from: https://www.alpha1.org/newly-diagnosed/learning-about-alpha-1/testing-for-alpha-1/
- American Lung Association [Internet]. Chicago: American Lung Association; c2023. Alpha-1 Antitrypsin Deficiency Symptoms and Diagnosis; [updated 2022 Nov 17; cited 2023 Jun 14]; [about 3 screens]. Available from: https://www.lung.org/lung-health-diseases/lung-disease-lookup/alpha-1-antitrypsin-deficiency/symptoms-diagnosis
- Cleveland Clinic: Health Library: Diseases & Conditions [Internet]. Cleveland (OH): Cleveland Clinic; c2023. Alpha-1 Antitrypsin Deficiency; [reviewed 2022 Oct 18; cited 2023 Jun 14]; [about 12 screens]. Available from: https://my.clevelandclinic.org/health/diseases/21175-alpha-1-antitrypsin-deficiency
- Merck Manual Consumer Version [Internet]. Kenilworth (NJ): Merck & Co. Inc.; c2023. Alpha-1 Antitrypsin Deficiency; [modified 2022 Sep; cited 2023 Jun 14]; [about 4 screens]. Available from: https://www.merckmanuals.com/home/lung-and-airway-disorders/chronic-obstructive-pulmonary-disease-copd/alpha-1-antitrypsin-deficiency
- National Heart, Lung, and Blood Institute [Internet]. Bethesda (MD): U.S. Department of Health and Human Services; Alpha-1 Antitrypsin Deficiency; [updated 2022 Mar 24; cited 2023 Jun 14]; [about 6 screens]. Available from: https://www.nhlbi.nih.gov/health-topics/alpha-1-antitrypsin-deficiency
- MedlinePlus [Internet]. Bethesda (MD): National Library of Medicine (US); What is a gene?; [cited 2023 Jun 14]; [about 3 screens]. Available from: https://medlineplus.gov/genetics/understanding/basics/gene/
- Testing.com [Internet]. Seattle (WA): OneCare Media; c2023. Alpha-1 Antitrypsin Testing; [modified 2021 Dec 13; cited 2023 Jun 14]; [about 13 screens]. Available from: https://www.testing.com/tests/alpha-1-antitrypsin/
- Testing.com [Internet]. Seattle (WA): OneCare Media; c2023. Bilirubin Test; [modified 2022 Nov 29; cited 2023 Jun 14]; [about 10 screens]. Available from: https://www.testing.com/tests/bilirubin/
- Testing.com [Internet]. Seattle (WA).: OneCare Media; c2023. Liver Panel; [cited 2023 July 5]; [about 1 screen]. Available from: https://www.testing.com/tests/alpha-1-antitrypsin/
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.
Health Library Alpha-1 Antitrypsin Deficiency (AATD)
What is an alpha-1 antitrypsin deficiency (aatd).
Alpha-1 antitrypsin (ān'tē-trĭp'sĭn) deficiency (AATD) is a disease that is passed down from parents to children. It can cause liver and lung disease. The liver makes a protein called alpha-1 antitrypsin that goes into the bloodstream. This protein protects the lungs and allows them to work normally. If there is not enough alpha-1 antitrypsin available in the body, it is called alpha-1 antitrypsin deficiency (AATD).
AATD is the most common genetic cause of liver disease in children. It is the most common genetic disease that leads to liver transplant in children.
Causes of Alpha-1 Antitrypsin Deficiency
Alpha-1 antitrypsin is a protein that is made in the liver and then released into the bloodstream. In AATD, the body does not make the correct form of this protein. Our bodies need alpha-1 antitrypsin to protect tissues in the body, particularly the lungs. The lack of this protein in the lungs leads to lung disease during adult years. At the same time, the build-up of the abnormal protein in the liver leads to liver disease. About 10% of patients with the severe form of AATD have liver disease that eventually requires a liver transplant.
AATD is an inherited condition and does not appear unless a person receives the same defective gene from both parents. If both parents carry an abnormal gene for AATD there is:

- A 25% chance their child will have the disease
- A 50% chance their child will receive one abnormal gene from one of the parents, which means the child will not show symptoms of the disorder but is a "carrier"
- A 25% chance their child will receive both normal genes, one from each parent, and will be unaffected
It is important to recognize that the severity of the liver and lung disease linked with AATD can vary greatly. Many people with AATD may have no or minimal liver involvement.
Signs and Symptoms
- Jaundice (or yellowing of the skin and eyes) that does not resolve
- Dark urine or pale stools
- Itching of the skin
- Enlarged liver
- Fluid build-up in the belly (called ascites)
- Trouble eating
- Poor growth or failure to thrive
- Elevated liver enzyme levels
Other children may not show signs of this condition until early childhood. First signs at this age may include:
- Easily tired
- Loss of appetite
- Severe itching of the skin
- Panniculitis (rare form of skin disease that causes the skin to harden and form lumps)
- Enlargement of the liver or spleen
Diagnosis of Alpha-1 Antitrypsin Deficiency
AATD is diagnosed with a simple blood test that measures the type of alpha-1 antitrypsin found in the blood. This test can tell whether a person has AATD or is a carrier. The blood test can be done soon after a baby is born if there is a family history of the deficiency. A test also is available to check if a baby in the womb has the condition.
Liver disease from AATD is diagnosed through a physical exam and abnormal changes found in the blood. A doctor can tell by feel during a physical exam if something is not normal. Tests, such as an ultrasound of the liver and spleen and a liver biopsy , can help confirm the diagnosis.
If bilirubin (a liquid produced in the liver that removes toxins from the body and helps break down fat in food) levels are higher than normal, it may be a sign of AATD. Increased levels of certain enzymes and abnormal ratios of certain proteins may also indicate liver disease.
Treatment for Alpha-1 Antitrypsin Deficiency
Alpha-1 antitrypsin deficiency affects children differently. One child may not show any signs of liver disease and another child may be seriously affected. Overall, only a small percentage of children develop liver disease related to AATD.
There is no cure for AATD. If severe liver disease develops, a liver transplant is currently the only option available for survival. The goal of treatment is to relieve the symptoms.
- Medicine may be given for severe itching.
- Diuretics (medicines that help remove excess fluid in the body) may be used to help reduce the buildup of fluid in the belly.
- A healthy diet and vitamin supplements can provide needed nutrients and may increase overall health.
- Vitamin/nutrition supplements may increase the effectiveness of the digestive process and increase energy levels.
Transplantation results in a cure of AATD. If a transplant is the best treatment option, the doctor and the other members of the patient care team focus on preventing complications. They will treat symptoms while your child waits for a donated liver. A liver transplant totally replaces the abnormal liver cells that produce the abnormal deficiency and corrects the protein abnormality.
It is critical for a child with AATD to avoid smoking or being exposed to second-hand smoke.
Long-Term Outlook
There is good chance of avoiding liver disease since only about 10% of children with alpha-1-antitrypsin deficiency develop significant liver disease. Liver transplants have been effective in reversing the symptoms of liver failure due to alpha-1 antitrypsin deficiency.
Related Resource
- Alpha-1 Foundation
Last Updated 01/2024
Connect With Us
3333 Burnet Avenue, Cincinnati, Ohio 45229-3026
© 1999-2024 Cincinnati Children's Hospital Medical Center. All rights reserved.

Learn how UpToDate can help you.
Select the option that best describes you
- Medical Professional
- Resident, Fellow, or Student
- Hospital or Institution
- Group Practice
- Patient or Caregiver
- Find in topic
RELATED TOPICS
INTRODUCTION
The pulmonary manifestations, diagnosis, and natural history of this disorder will be reviewed here [ 1-4 ]. Extrapulmonary disease and therapy are discussed separately. (See "Extrapulmonary manifestations of alpha-1 antitrypsin deficiency" and "Treatment of emphysema from alpha-1 antitrypsin deficiency" .)
Emphysema in AAT deficiency (AATD) is thought to result from an imbalance between neutrophil elastase in the lung, which destroys elastin, and the elastase inhibitor AAT, which is synthesized in hepatocytes and protects against proteolytic degradation of elastin [ 4 ]. This mechanism is called a "toxic loss of function." Specifically, cigarette smoking and infection increase the elastase burden in the lung, thus increasing lung degradation [ 1 ]. In addition, the polymers of "Z" antitrypsin are chemotactic for neutrophils, which may contribute to local inflammation and tissue destruction in the lung [ 7 ].
The pathogenesis of the liver disease is quite different and is called a "toxic gain of function." The liver disease results from the accumulation within the hepatocyte of unsecreted variant AAT protein. Only those genotypes associated with pathologic polymerization of AAT within the endoplasmic reticulum of hepatocytes (eg, PI*ZZ type AATD) produce disease [ 8-10 ]. Most patients with liver disease due to AATD are homozygous for the Z allele (ie, PI*ZZ); liver disease does not occur in null homozygotes who have severe deficiency of AAT, but no intra-hepatocytic accumulation. (See "Extrapulmonary manifestations of alpha-1 antitrypsin deficiency", section on 'Hepatic disease' .)
Alpha-1 Antitrypsin Deficiency
What is alpha-1 antitrypsin deficiency (alpha-1).
Alpha-1 is a genetic disorder that affects the lungs and sometimes the liver. Even though it is one of the most common genetic disorders, Alpha-1 can be hard to diagnose. One challenge is that most people with Alpha-1 are healthy for at least the first few decades of their lives. For many, symptoms do not appear until middle adulthood. Another challenge is that the effects of the disorder look a lot like other conditions. Lung symptoms can mimic asthma, bronchitis, or smoking-induced emphysema. Liver symptoms can mimic cirrhosis. This often leads to misdiagnosis and a delay in treatment. In the United States, more than 90% of people with Alpha-1 never learn that they have it.
Affected gene
The affected gene in Alpha-1 is SERPINA1, on chromosome 14. This gene codes for a protein called alpha-1 antitrypsin (AAT). People with the disorder have two non-working copies (alleles) of the gene; they make little or no working AAT protein.
AAT protein is normally made in the liver and released into the blood stream. From there, it can travel throughout the body—most importantly to the lungs. When we breathe in irritants like viruses or smoke, AAT protects the lungs from damage.
In people with Alpha-1, very little or no AAT protein makes it to the lungs. The lungs are left unprotected. Some people who have Alpha-1 make a sticky version of AAT protein that builds up in the liver. Not only do their lungs become damaged, but the sticky AAT protein can also harm the liver.
People with Alpha-1 inherit two non-working copies (alleles) of the SERPINA1 gene: one from each parent. Not everyone with two non-working alleles will develop the symptoms of Alpha-1. Yet they are still considered to have a genetic disorder because their AAT levels are low, or deficient.

Alpha-1 antitrypsin protein is active in the lungs, where it protects them from damage. Without the protein, the lungs slowly accumulate damage over the course of a lifetime.
Inheritance
From the perspective of having the genetic disorder, Alpha-1 mostly follows an autosomal recessive inheritance pattern: it takes two non-working alleles to cause the disorder.
From the perspective of the amount of AAT protein that is made, a person's two SERPINA1 alleles are co-dominant: protein is made from both.

Protein function and interactions

AAT protein inactivates elastase. Without AAT protein, elastase attacks stretchy proteins in lung tissue, causing damage to the lungs.
The most important job of alpha-1 antitrypsin (AAT) protein is to protect the lungs from a protein called neutrophil elastase.
Elastase is made naturally by immune cells. When we breathe in irritants—things like dust, smoke, viruses, or chemicals—immune cells travel to the lungs and begin releasing elastase. Elastase breaks down invading bacteria and viruses, thus protecting us from infection.
If no AAT protein is present, elastase will also attack a structural protein called elastin. In the lungs, elastin is essential for helping tissue expand and contract as we breathe. As elastin is broken down, holes form in delicate lung tissue, making breathing difficult.
Normal protein expression
The SERPINA1 gene is switched off in most cell types. SERPINA1 is active at a low level in kidney and immune cells, and at a high level in liver cells.
AAT protein does not normally stay inside of the cells that make it. Rather, it is released to the outside of the cells. Most AAT protein in the body comes from liver cells. They release AAT into the bloodstream, where it can travel to all of the tissues in the body—including the lungs.

Alpha-1 antitrypsin (AAT) protein is shown in purple. Most of the body's AAT protein is made by cells in the liver and released into the blood stream. Inside of liver cells, AAT protein is found in the endoplasmic reticulum and Golgi apparatus, and it is secreted to the outside of the cell.
Symptoms and features of Alpha-1

Many people with Alpha-1 develop emphysema (left). The tiny air sacs in the lungs, called alveoli, are damaged. This makes the air spaces in the lungs larger, decreases the surface area of the lungs, and makes breathing difficult. Some people with Alpha-1 make a version of AAT protein that builds up inside of liver cells (right).
The effects of Alpha-1 can be very different from person to person. The effects depend mostly on the specific gene variants (alleles) that a person has.
The main feature of Alpha-1 is damage to the tiny air sacs (alveoli) in the lungs. When the alveoli are damaged, the lungs have a harder time taking in oxygen. This can cause shortness of breath, coughing, or wheezing. Over time, people with Alpha-1 may develop lung diseases, such as asthma, emphysema, chronic bronchitis, or COPD (Chronic Obstructive Pulmonary Disease). Lung disease can appear as early as age 30. These conditions are life-threatening, with symptoms that get worse over time.
Certain SERPINA1 gene variants (alleles) can also cause liver damage. These alleles code for a sticky version of AAT protein that builds up in liver cells. Liver problems include liver cancer, liver failure, and cirrhosis (a chronic liver disease). Liver symptoms can appear at any time, affecting infants, children, and adults.
In rare cases, hard and painful lumps may form under the skin. This is called panniculitis.
Alleles, protein, and variability
The gene variants, or alleles, that a person has directly affects the characteristics of the AAT protein a person makes, including how much working protein is released into the blood. It's not a simple matter of whether a person makes AAT protein or not. Rather, AAT protein levels vary across a spectrum, from high to low. To further complicate matters, our AAT levels can change over time. They tend to go up when we are sick or stressed and down when we are healthy.
The amount and type of AAT protein that a person makes impacts their risk for lung and liver disease. In general, the less AAT protein in a person's blood, the higher their risk for lung damage.
The amount of AAT protein in a person's blood can be measured using a simple blood test. If a blood test shows that a person has low levels of AAT, the usual follow up is to do genetic testing, along with genetic counseling. Genetic testing can reveal which SERPINA1 alleles the person has. Each allele combination comes with its own set of risks.
Among people diagnosed with alpha-1, the ZZ allele combination is the most common. About 75% of people with the ZZ allele combination develop lung disease, especially later in life. People with the ZZ allele combination also have a high risk for liver disease, which can appear at any time (the percentage who will develop liver symptoms is unknown).

AAT protein levels vary across a spectrum. In this hypothetical graph, each point represents the concentration of working AAT protein from one person at one time point. Their allele combinations are shown on the X-axis.
Allele categories: M = "healthy"; S = reduced AAT function; Z = "sticky" AAT protein with very low function; Null = essentially no AAT protein. Data based on Ferrarotti et al.
Other Factors

Differences in genes and exposure to different environmental factors accounts for many of the trait differences among family members.
Even among people who have the same SERPINA1 alleles, there is a lot of individual variation. People with the same alleles can differ in the amount of AAT protein that they make, and in the timing and types of symptoms that they develop. Even among family members, some may have symptoms at a young age, and others may be symptom-free for life.
This variability is due to environmental factors and variations in other genes. Environmental factors include things like exposure to cigarette smoke, and the viruses a person has as a child. Gene interactions include variations in genes that influence things like how much elastase white blood cells make, or how well liver cells can clear protein clumps that build up inside of them—plus variations in other genes that influence a person's risk for lung disease.
Treating and managing Alpha-1
To manage their health, people with Alpha-1 can use a combination of lifestyle behaviors and medical approaches. Lifestyle behaviors are aimed at keeping the body as strong and healthy as possible. Medical approaches are aimed at preventing and managing infections, and easing symptoms.
Carriers of one disease-causing allele may have a higher-than-average risk for lung and liver disease. But usually it also takes an unhealthy environmental factor—such as obesity or cigarette smoking—for disease to develop. Carriers should follow the recommended lifestyle behaviors below and stay up-to-date on their vaccinations. Carriers should also talk to their doctors if they see warning signs of disease.
Lifestyle behaviors
- Limit tobacco smoke and air pollution. Lung irritants cause inflammation (and increase levels of neutrophil elastase) and block the protective effects of AAT.
- Develop an exercise program. A stronger body has more stamina.
- Limit drinking alcohol. This is especially important in people who have alleles that can affect the liver.
- Eat a nutritious diet.
- Limit stress.

Tobacco smoke is especially harmful to people with Alpha-1.
Medical approaches
- Vaccinate against influenza, pneumonia, and hepatitis. These preventable illnesses can damage the lungs or liver.
- Aggressively treat lung infections. This can decrease inflammation and prevent lung damage.
- Asthma medications, such as bronchodilators, can ease lung symptoms.
- Corticosteroids can reduce lung inflammation.
- Supplemental oxygen.
- Protein therapy (also called augmentation therapy) can help to slow lung damage. It involves injecting working AAT protein into the blood stream.
- Lung surgery can be helpful in some cases.
- In later stages of disease, if the lungs or liver become heavily damaged, people with Alpha-1 may receive an organ transplant.
Alpha-1 is a potential target for genetic technology. One approach is gene therapy, in which a modified virus delivers a working copy of the SERAPINA1 gene into the patient's cells.

Vaccinating against preventable illnesses can help to protect the lungs and liver.
More information
The Alpha-1 Foundation provides confidential testing and genetic counseling to the Alpha-1 community. It is also an excellent resource both for the public and for health care professionals.
The Alpha-1 Foundation (December 2015). Alpha-1 Antitrypsin Deficiency Healthcare Provider's Guide, version 2.0. Accessed 3/10/2018 at https://www.alpha1.org
Ferrarotti, I., Thun, G. A., Zorzetto, M., Ottaviani, S., Imboden, M., Schindler, C., ... & Probst-Hensch, N. M. (2012). Serum levels and genotype distribution of α1-antitrypsin in the general population. Thorax, 67(8), 669-674.
Lazarin, G. A., Haque, I. S., Nazareth, S., Iori, K., Patterson, A. S., Jacobson, J. L., ... & Srinivasan, B. S. (2013). An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genetics in Medicine, 15(3), 178.
Marvel, J., Yu, T. C., Wood, R., Higgins, V. S., Make, B. J., Sandhaus, R. A., ... & Goodman, K. (2016). The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstructive Pulmonary Diseases: Journal of the COPD Foundation, 3(3), 668.
- Help & Support
- Lung Health & Diseases
- Lung Disease Lookup
- Alpha-1 Antitrypsin Deficiency
Alpha-1 Antitrypsin Deficiency Symptoms and Diagnosis
What are the symptoms of aat deficiency.
Symptoms can appear early in life, but many symptoms will not begin until a person reaches middle-age. People with AAT deficiency may have a wide variety of breathing-related symptoms like:
- Shortness of breath
- Chronic cough with sputum (mucus or phlegm) production
- Reduced exercise ability
- Fatigue or tiredness
- Frequent lung infections like cold or flu
When the liver is affected by AAT deficiency, symptoms may include tiredness, loss of appetite, weight loss, swelling of the feet or belly, yellowish discoloration of the skin (jaundice) or white part of the eyes, vomiting of blood, or blood in stools.
In rare cases, AAT can cause a skin disease called panniculitis, resulting in hardened patches and red, painful lumps.
Early diagnosis of AAT deficiency can help prevent COPD from developing.
How AAT Deficiency Is Diagnosed
AAT deficiency runs in families so if you have family members with AATD, developed COPD in your 40s or 50s, or have liver disease, it is important medical history to discuss with your healthcare provider. Your healthcare provider may order you a blood test to check the level of AAT protein in your bloodstream. People who smoke with AAT deficiency tend to develop disease 10 or more years earlier than people who do not smoke.
Global Initiative for Chronic Obstructive Lung Disease (GOLD) Guidelines recommend that all people with COPD, regardless of age or ethnicity, should be tested for AAT deficiency. If you have a close family member—such as a parent or sibling—with AAT deficiency you should also be screened. Tests and procedures for healthcare provider may perform include:
- Blood test to check the level of alpha-1 antitrypsin protein in your body. If your levels are low, genetic testing with another blood test may be used to identify any abnormal genes.
- Lung function tests like a spirometry test cannot diagnose AAT deficiency but it can tell how well a person’s lungs are working.
- Imaging tests like a chest X-ray or CT scan of your lungs can check for damage in the lungs or rule out other conditions.
If your healthcare provider suspects AAT deficiency is affecting the liver, the provider may order blood testing of liver function and in some cases an ultrasound of the liver. If you have low levels of AAT but normal liver and lung function tests, you may not need treatment; however, you will be monitored with repeat testing over time.
When to See Your Doctor
Reviewed and approved by the American Lung Association Scientific and Medical Editorial Review Panel
Page last updated: June 7, 2024
A Breath of Fresh Air In Your Inbox
Join over 700,000 people who receive the latest news about lung health, including research, lung disease, air quality, quitting tobacco, inspiring stories and more!
Thanks for submitting your email.
Make a Donation
Your tax-deductible donation funds lung disease and lung cancer research, new treatments, lung health education, and more.
Become a Lung Health Insider
Thank you! You will now receive email updates from the American Lung Association.
Select Your Location
Select your location to view local American Lung Association events and news near you.
Change Language
Lung helpline.
Talk to our lung health experts at the American Lung Association. Our service is free and we are here to help you.
1-800-LUNG-USA
(1-800-586-4872)
IMAGES
VIDEO
COMMENTS
Alpha-1 is a rare disease that makes an enzyme in your liver work poorly. Alpha-1 antitrypsin protein usually travels from your liver through your blood to protect your lungs and other organs. But ...
Description. Alpha-1 antitrypsin deficiency is an inherited disorder that may cause lung disease and liver disease. The signs and symptoms of the condition and the age at which they appear vary among individuals. People with alpha-1 antitrypsin deficiency usually develop the first signs and symptoms of lung disease between ages 25 and 50.
Alpha-1-antitrypsin (AAT) is a protein produced by the liver that protects the lungs from inflammation and damage caused by inhaling irritants such as tobacco or wildfire smoke. It is estimated that there are between 80,000 to 100,000 people living with AAT deficiency in the United States, putting them at greater risk for developing COPD.
Alpha-1 Antitrypsin Deficiency. Alpha-1 antitrypsin (AAT) deficiency is a rare genetic disorder that is passed on in families and can affect the lungs, liver and/or skin. When this condition affects the lungs, it causes COPD (chronic obstructive pulmonary disease).
Alpha-1 antitrypsin deficiency (AAT deficiency, or AATD) is an inherited condition that raises your risk for lung and liver disease. If you have this condition, your body doesn't make enough alpha-1 antitrypsin (AAT). AAT is made by your liver. It helps protect your lungs from inflammation and irritating substances you might breathe in, such as ...
Alpha-1 antitrypsin deficiency (A1AD or AATD) is a genetic disorder that may result in lung disease or liver disease. Onset of lung problems is typically between 20 and 50 years of age. This may result in shortness of breath, wheezing, or an increased risk of lung infections. Complications may include chronic obstructive pulmonary disease (COPD), cirrhosis, neonatal jaundice, or panniculitis.
Alpha-1 antitrypsin deficiency (AATD) is an inherited condition that causes low levels of, or no, alpha-1 antitrypsin (AAT) in the blood. AATD occurs in approximately 1 in 2,500 individuals. This condition is found in all ethnic groups; however, it occurs most often in whites of European ancestry. Alpha-1 antitrypsin (AAT) is a protein that is ...
Alpha-1 antitrypsin (AAT) deficiency is a genetically inherited disorder often unrecognized in clinical practice. It results in impaired production of alpha-1 antitrypsin protein, which plays a role in protecting the body from neutrophil elastase, an enzyme released by white blood cells during infection. Due to defective protein production, there is reduced activity of AAT in the blood and lungs.
Alpha-1 antitrypsin (AAT) deficiency increases an individual's risk for COPD. The deficiency is an inherited genetic condition with no cure. The resulting COPD would be treated in the standard manner, with bronchodilators, steroids, pulmonary rehabilitation, oxygen therapy, and surgery. AAT is diagnosed with a blood test.
Alpha-1 antitrypsin deficiency (AATD) can present with hepatic dysfunction in individuals from infancy to adulthood and with chronic obstructive lung disease (emphysema and/or bronchiectasis), characteristically in individuals older than age 30 years. Individuals with AATD are also at increased risk for panniculitis (migratory, inflammatory, tender skin nodules which may ulcerate on legs and ...
Alpha-1 Antitrypsin Deficiency. Reviewed/Revised May 2024. Alpha-1 antitrypsin deficiency is a hereditary disorder in which a lack or low level of the enzyme alpha-1 antitrypsin damages the lungs and liver. Alpha-1 antitrypsin deficiency is caused by an inherited gene mutation. Infants may develop jaundice and liver damage.
Alpha-1 antitrypsin deficiency (sometimes just called "Alpha-1") is an inherited genetic disorder that causes low levels of a protein (AAT) that protects your lungs. Alpha-1 increases your risk of developing certain diseases, including emphysema (damaged air sacs in your lungs), cirrhosis (liver scarring) and panniculitis (an uncommon skin ...
More information about AAT deficiency is available from the Alpha-1 Foundation, such as a nationwide network of affiliated support groups for alpha-1 patients and families. Finding Support. Communicate regularly with your doctors about changes in your breathing and general health. The Lung Association recommends patients and caregivers join our ...
Understanding Alpha-1 Alpha-1 is an inherited condition that is present at birth. Alpha-1 may result in serious lung disease in adults and/or liver disease at any age. Approximately 100,000 people in the United States are estimated to have Alpha-1. In typical individuals, large amounts of the alpha-1 antitrypsin (AAT) protein are made in the liver
If you have AAT deficiency, the counselor can provide information on the condition, including your risk of passing the disease to your children. Alpha-1 antitrypsin (AAT) is a protein that protects the lungs and liver from damage. AAT testing is used to diagnose a condition called AAT deficiency.
Alpha-1 antitrypsin (ān'tē-trĭp'sĭn) deficiency (AATD) is a disease that is passed down from parents to children. It can cause liver and lung disease. The liver makes a protein called alpha-1 antitrypsin that goes into the bloodstream. This protein protects the lungs and allows them to work normally. If there is not enough alpha-1 ...
Alpha-1 antitrypsin deficiency, or Alpha-1, is a rare genetic disorder that runs in certain families and that most often affects the lungs and liver. Approximately 70,000 to 100,000 Americans may have the disorder, though most have not been diagnosed. Diagnosis of Alpha-1 is based on a laboratory blood test.
INTRODUCTION. Alpha-1 antitrypsin (AAT) deficiency is a clinically under-recognized inherited disorder affecting the lungs, liver, and rarely, skin. In the lungs, AAT deficiency causes chronic obstructive pulmonary disease (ie, emphysema and bronchiectasis). The pulmonary manifestations, diagnosis, and natural history of this disorder will be ...
Alpha-1 antitrypsin deficiency (A1AD) is a hereditary disorder characterized by low levels of a protein called alpha-1 antitrypsin (A1AT) which is found in the blood. This deficiency may predispose an individual to several illnesses and most commonly manifests as chronic obstructive pulmonary disease (including bronchiectasis) and liver disease ...
What is Alpha-1 Antitrypsin Deficiency (Alpha-1)? Alpha-1 is a genetic disorder that affects the lungs and sometimes the liver. Even though it is one of the most common genetic disorders, Alpha-1 can be hard to diagnose. One challenge is that most people with Alpha-1 are healthy for at least the first few decades of their lives.
Shortness of breath. Chronic cough with sputum (mucus or phlegm) production. Wheezing. Reduced exercise ability. Fatigue or tiredness. Frequent lung infections like cold or flu. When the liver is affected by AAT deficiency, symptoms may include tiredness, loss of appetite, weight loss, swelling of the feet or belly, yellowish discoloration of ...
Alpha-1 is a condition that affects the lungs and/or the liver. It is a progressive condition, which means it may worsen over time. It can lead to serious lung and/or liver disease. Alpha-1 is also known as AAT, AATD, Alpha-1 protease inhibitor deficiency, Alpha-1 related emphysema, genetic emphysema, hereditary pulmonary emphysema, inherited ...
Alpha-1 Foundation Toll Free: (877) 228-7321 • alpha1.org The not-for-profit Foundation provides resources, education and information on testing and diagnosis for healthcare providers and people affected by Alpha-1. It funds cutting-edge research to find treatments and a cure, and supports worldwide detection of Alpha-1. AlphaNet